Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Pharmacometabolomics wikipedia , lookup
Basal metabolic rate wikipedia , lookup
Light-dependent reactions wikipedia , lookup
Nicotinamide adenine dinucleotide wikipedia , lookup
Microbial metabolism wikipedia , lookup
Electron transport chain wikipedia , lookup
NADH:ubiquinone oxidoreductase (H+-translocating) wikipedia , lookup
Evolution of metal ions in biological systems wikipedia , lookup
Mitochondrion wikipedia , lookup
Lactate dehydrogenase wikipedia , lookup
Fatty acid metabolism wikipedia , lookup
Adenosine triphosphate wikipedia , lookup
Oxidative phosphorylation wikipedia , lookup
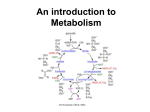
Citric acid cycle wikipedia , lookup
Glyceroneogenesis wikipedia , lookup
Blood sugar level wikipedia , lookup
NUTR 6104 Melissa Paul 2013 Fall Exam 1 October 17, 2013 (Due October 31, 6:00 PM) You are expected to work independently (no discussion with class mates or others not in class). You have open access to information. Write in your own words. Drop at the Dropbox of D2L. I. Please answer the following questions (15 points). 1. What is the function of the malate shuttle? The malate shuttle moves electrons produced during glycolysis across the semi permeable inner membrane of the mitochondrion for oxidative phosphorylation. These protons enter the electron transport chain of the mitochondria via reduction equivalents to generate ATP. The shuttle system is required because the mitochondrial inner membrane is impermeable to NADH, the primary reducing equivalent of the electron transport chain. To move NADH from the cytosol to the mitochondrial matrix the malate shuttle is required. Malate carries the reducing equivalents across the membrane. 2. Where the electron transport from NADH and FADH2 to O2 occurs in. The electron transport chain is located on the inner mitochondrial membrane. NADH enters at Complex I and FADH2 enters at Complex II; protons (H+) are passed to O2 at Complex IV to form H2O. 3. Energy for the transport of ADP into the mitochondria matrix. (Where is the energy coming from for the transport of ADP into the mitochondria matrix?) Oxidative phosphorylation, which takes place during the process of cellular respiration, in addition to the substrate‐level phosphorylation, or phosphorylation o f ADP to ATP by the transfer of a high‐energy phosphate bond to a compound, which occurs during glycolysis and the Krebs cycle. During oxidative phosphorylation, NADH is oxidized to NAD+, yielding 2 ATPs, and FADH2 yields 1 ATP when oxidized. Oxidative phosphorylation uses the gradient of protons (H+) pumping across the inner mitochondrial membrane to generate ATP from ADP. 4. In which tissue, the conversion of glucose 6‐phosphate to lactate is a major pathway for ATP production. Glucose 6‐phosphate never goes directly to lactate; however, under anaerobic conditions, such as exercising skeletal muscle and in RBCs, the metabolic process of 1 glucoseglucose 6‐phosphate pyruvate will then process pyruvate to lactate (reversed by the Cori Cycle). 5. Glucokinase and hexokinase convert glucose to glucose 6‐phosphate. What enzyme is involved in the reverse reaction? Glucose 6‐phosphatase 6. A step in glycolysis that is positively regulated by the concentration of the allosteric regulator, fructose‐2,6‐bisphosphate. Allosteric regulation at Fructose 6‐P Fructose 1,6‐bis‐P (catalyzed by phosphofructokinase) 7. List two major tissues express glucose 6‐phosphatase, fructose‐1,6‐ bisphosphatase, pyruvate carboxylase, and phosphoenolpyruvate carboxykinase. Liver and to lesser extent in kidney 8. If the cytosolic NADH/NAD+ ratio is pushed to very high levels, such that NAD+ is not readily available to the cell (NAD+ is low), pyruvate will preferably be converted to A molecule. What is A molecule? Lactate 9. What enzyme in anabolic carbohydrate metabolism is inactive when phosphorylated? Glycogen synthase 10. The maximum numbers of ATP equivalents that can be obtained by glucose metabolism by a red blood cell and liver cell, respectively. No O2 available or no mitochondria (RBC): Glucose must be converted to lactate, 2 ATP produced Liver (O2 available): maximum ATP = 38 ATP II. Please answer the following questions (15 points). 1. What is the role of substrate‐level phosphorylation in glycolysis? 2 Substrate‐level phosphorylation is the direct transfer and donation of a phosphoryl (PO3) group to adenosine diphosphate (ADP). Specifically in glycolysis there are two steps where substrate‐level phosporylation occurs: Two molecules of 1,3‐biphosphoglycerate are converted to 3‐phosphoglycerate, and the high‐energy phosphate group is added to ADP producing 2 ATP by substrate‐level phosphorylation. The two molecules of 3‐phosphoglycerate are rearranged to form two molecules of 2‐phosphoglycerate; hydrolysis of the two molecules of 2‐phosphoglycerate occurs, converting the phosphate bonds to a high‐energy phosphate bonds with two molecules of phosphoenolpyruvate produced. As the two molecules of phosphoenolpyruvate are converted to two molecules of pyruvate, the high‐ energy phosphate groups are added to ADP producing 2 ATP by substrate‐level phosphorylation. 2. Under what conditions is glucose converted to lactic acid by skeletal muscle? Anaerobic conditions 3. How does pyruvate, the carbon‐containing product of glycolysis, enter the mitochondria and what happens to this molecule once inside the mitochondria? Pyruvate enters the mitochondria by active transport. Once inside the matrix of the mitochondria pyruvate must be converted into Acetyl‐CoA by pyruvate dehydrogenase in combination with CoASH and NAD+ to enter the Kreb's Cycle. CO2 and NADH, H+ are also products of this reaction. 4. What comprises the proton‐motive force in mitochondria? The proton‐motive force in mitochondria generates ATP by the movement of hydrogen ions down their electrochemical gradient, across a membrane during cellular respiration. Specifically, hydrogen ions (H+, protons) that originate from NADH and FADH2 diffuse from an area of high concentration to an area of lower concentration. ATP synthase is the enzyme that makes ATP by chemiosmosis. It allows protons to pass through the membrane and uses the kinetic energy to phosphorylate ADP, making ATP. The generation of ATP by chemiosmosis occurs in mitochondria. 5. In normal individuals, the capacity of the liver to phosphorylate fructose (fructokinase activity) greatly exceeds the liver’s capacity to split fructose 1‐ phosphate (adolase B activity). Why is deficiency of fructokinase a less serious genetic defect than a deficiency of fructose 1‐phosphate adolase? Consider what happens to fructose in each case and what effect this has on hepatic metabolism. 3 Fructose is converted to fructose‐1‐phosphate in the liver by fructokinase. Fructokinase is classified as a transferase, specifically a phosphotranferase, because its role is to transfer a phosphate group. Fructokinase is needed for the synthesis of glycogen from fructose. Deficiency of fructokinase is a less serious genetic defect that causes only fructosuria, or high levels of fructose in the blood, with the fructose being excreted unchanged in the urine. The following step of fructose degradation, fructose‐1‐phosphate, relies on aldolase B to split the six carbon chain into dihydroxyacetone phosphate and glyceraldehyde. A deficiency of aldolase B is a more serious genetic defect that results in an accumulation of fructose‐1‐phosphate, and trapping of phosphate. Aldolase B is essential in carbohydrate metabolism as it catalyzes this major step of glycolysis and gluconeogenesis. This enzyme is particularly important for fructose metabolism. Genetic mutations leading to a lack of functional aldolase B result in a condition called hereditary fructose intolerance. F1P accumulation is toxic to cellular tissues and traps phosphate in an unusable form that causes depletion of both phosphate and ATP stores. The lack of readily available phosphate down regulates glycogenolysis in the liver, which results in hypoglycemia. F1P accumulation also inhibits gluconeogenesis, further reducing the amount of readily available glucose. The loss of ATP leads to a multitude of problems including inhibition of protein synthesis and hepatic and renal dysfunction. However, hereditary fructose intolerance can be managed through avoidance of foods containing fructose, sucrose, and sorbitol. 6. If your friend has problems in producing NADH, what life style do you suggest for your friend? ↓ATP produc on Problems producing NADH would imply a niacin deficiency. I would suggest this friend lead a somewhat sedentary lifestyle due to low energy production. III. Discuss the following questions. (45 points) A. Carbohydrate intolerance is a rare but very serious hereditary disorder. Cases due to lack of digestive enzyme (e.g., primary sucrase‐ dextrinase deficiency) and to impaired hexose transport (i.e., primary glucose‐galactose malabsorption) have been reported. In two cases of carbohydrate intolerance, the infants developed diarrhea soon after birth, and reducing sugars and an acid pH were present in the stools. Intestinal biopsy tissues from the infants showed normal villi with normal disaccharidase values. Monosaccharide load tests showed normal rise in blood glucose in response to a fructose load (=not a sucrose problem), which was accompanied by marked abdominal distention, profuse diarrhea, and reducing sugars in the stools. Thus these two patients were diagnosed as having glucose‐ galactose malabsorption. 4 Although a sucrose‐ and lactose‐free formula with fructose was fed to these two patients, diarrhea and the excretion of reducing sugars in the stool continued. Rigid exclusion of all carbohydrates and sugars except fructose resulted in cessation of diarrhea and elimination of sugars from the stools. 1. Which hexose transporter protein was probably lacking in these patients? SGLT1 2. If the plasma glucose concentration had been monitored in these two patients following a load dose of sucrose, would a rise in plasma glucose have been observed? How about a following a load of lactose? ‐2.5 Yes, a rise would occur. SGLT1 would be required after sucrase enzyme breaks sucrose down to monosaccharides, glucose and fructose. The glucose transporter, SGLT1, is lacking to bring glucose into the cell. Yes, a rise would occur. SGLT1 would be required after lactase enzyme breaks lactose down to monosachharides, glucose and galactose. SGLT1 is required for both to be brought into the cell. 3. How would you expect the findings (carbohydrate tolerance, the glucose load test results, and the intestinal biopsy results) to differ in a case of secondary disaccharidase deficiency due to infective gastroenteritis (e.g., rotavirus infection)? With disaccharidase deficiency disaccharides will not be broken to monosaccharides, and carbohydrate intolerance will occur. Therefore, glucose load test will not show elevated levels as the undigested sugars are fermented and pass directly to the colon. Symptoms such as nausea, bloating and diarrhea are likely to occur. Disaccharidases occur in the microvillus of the small intestine and lack of absorption causes atrophy of the villus from lack of use. ‐1.5 B. A rare inborn error of metabolism, or genetic defect, is congenital sucrose‐ isomaltase (sucrase‐ ‐dextrinase) deficiency. 1. What reactions in human digestion are catalyzed by this protein? Sucrose‐isomaltose ‐‐‐(sucrose‐isomaltase) 3 glucose + fructose Sucrose‐isomaltose is a bifunctional enzyme: sucrase breaks down sucrose; isomaltase cleaves alpha 1‐6 branching links of alpha‐limit dextrins. Sucrose‐isomaltase, or sucrase‐ alpha‐dextrinase, deficiency creates an inability to break down sucrose and starch due to a lack of the bifunctional enzyme sucrose‐isomaltase that hydrolyzes sucrose and isomaltose to glucose and fructose. 5 2. Would you expect patients with sucrase‐a‐dextrinase deficiency to have problems digesting (a) sucrose? (b) lactose? (c) maltose? (d) starch? Explain. (a) sucrose – yes (b) lactose – no (c) maltose – no (d) starch – yes As was also explained in the previous question, sucrose‐isomaltase, or sucrase‐alpha‐ dextrinase, splits into two enzyme substrate units, sucrase and isomaltase. Sucrase acts on sucrose and isomaltase acts to cleave alpha 1‐6 branching links. A patient with sucrose‐isomaltase deficiency has an inability to break down sucrose and starch due to a lack of this bifunctional enzyme that hydrolyzes sucrose and isomaltose into glucose and fructose. Lactase and maltase break down lactose and maltose respectively. Assuming the patient has no deficiency in either of these enzymes in addition to a sucrose‐isolmaltase deficiency, s/he should have no issues digesting lactose and maltose. C. A lack of glucose 6‐phosphatase in liver and kidney causes Von Gierke’s disease (Type I glycogen storage disease). This is caused by a genetic defect in the glucose 6‐ phosphatase gene that occurs as an autosomal recessive trait in 1 of 200,000 people. 1. Explain the underlying metabolic basis for development of (a) hypoglycemia, (b) lactic acidosis, and (c) hypertriglyceridemia in patients with glycogen storage disease Type I. Von Gierke’s disease: glucose‐6‐phosphatase deficiency impairs ability of liver to produce free glucose from glycogen, results in severe hypoglycemia and increase in glycogen storage in liver and kidneys (a) Hypoglycemia occurs because the lack of properly functioning glucose‐6‐phospatase causes a build up glucose‐6‐phosphate (glu‐6‐P), which prevents conversion of glycogen back into glucose in the liver (allosteric regulation of glycogenolysis is negative). Therefore, glucose is not supplied sufficiently to the rest of the body during a fasting state. This quickly leads to hypoglycemia and an increase in glycogen stores in the liver and kidneys. (b) Gluconeogenesis is also driven by glucose‐6‐phosphatase (glu‐6‐Pase), and when there is a deficiency in glu‐6‐Pase gluconeogenesis is impaired (Glu‐6‐P cannot convert 6 to glucose). As glucose‐6 phosphate builds up it simultaneously inhibits conversion of lactate to pyruvate for entry into the GNG pathway. With impaired GNG lactic acid levels rise. Overall, lactate is a precursor for GNG and GNG is impaired, so rising lactic acid levels causing lactic acidosis. (c) An additional secondary effect of excess glu‐6‐P is that glu‐6‐P is then directed into production of triglycerides and exported for storage in adipose tissue as fat. Chronically low insulin levels that are a secondary effect of hypoglycemia amplify this production of triglycerides. Good! 2. What effect does treatments with glycogen storage disease type I by providing carbohydrate through the day have on the overproduction of lactate and triacylglycerol? Explain? As previously discussed, Von Gierke’s creates an inability for the body to maintain adequate blood glucose levels during fasting due to inhibition of glycogenolysis and gluconeogenesis pathways. Eating more frequent, small meals that provide consistent carbohydrates throughout the day is the primary treatment for prevention of hypoglycemia. Specifically foods high in glucose or starch (over fructose and galactose) are readily digested to glucose for immediate use, and GNG in the liver is not necessary as a source of glucose. Moreover, lactic acid production in the liver due to falling blood glucose levels during fasting is avoided (i.e., lactic acidosis is avoided). When your body has a steady, constant supply of glucose (even through the night would be required) insulin levels are not then chronically low due to hypoglycemia. In avoiding this secondary effect of hypoglycemia and maintaining consistent levels of insulin, production of triglycerides is not amplified. D. Early attempts to use fructose instead of glucose in total parenteral nutrition resulted in severe liver damage. What happened to fructose metabolism in the liver? ‐1.0 The hepatic metabolism of fructose differs greatly from that of glucose. Contrary to glucose, fructose is metabolized exclusively in the liver by fructokinase. ‐‐‐‐‐form fructose 1‐P‐‐use up inorganic phosphate,… impossible to make ATP by the liver‐‐‐‐‐↓ATP‐dependent pumps Glucose metabolism is regulated by the rate‐limiting enzyme phosphofructokinase, which is inhibited by ATP and citrate. Additionally, the hepatic conversion of glucose to pyruvate is regulated by insulin. In contrast, the conversion of fructose into triose‐ Phosphate is a rapid process independent of insulin. Fructose bypasses the conversion of glucose‐6‐phosphate to fructose 1,6‐bisphosphate that is regulated by phosphofructokinase. Essentially, the low Km of fructokinase for fructose, and the absence of negative feedback by ATP or citrate allows for continuous, rapid entry of fructose into the glycolytic pathway. Therefore, fructose can rapidly and without any control produce glucose, glycogen, lactate, pyruvate, and from these precursors 7 ultimately produce large amounts of triglycerides. High glucose levels may increase formation of glycerol‐3‐phosphate and accelerate hepatic triglyceride production. E. A patient complained of muscle cramps and was unable to perform strenuous exercise. He had elevated levels of creatine phosphokinase, adolase, and myoglobin in his blood. The release of enzymes and other proteins from muscle cells is indicative of muscle cell damage. He was diagnosed with Type V glycogen storage disease (McArdle’s disease), which is caused by an absence of muscle glycogen phosphorylase. 1. Would you expect glycogen to accumulate in the muscle off this patient? Why or why not? Yes, one would expect glycogen to accumulate in the muscle of this patient because myophosphorylase is not available to break down muscle glycogen to glucose‐1‐ phosphate. The enzyme would normally remove 1,4 glycosyl residues from outer branches of glycogen while adding inorganic phosphate to form glucose‐1‐phosphate. 2. Would you expect lactate to accumulate in the muscle or blood of this patient during exercise? Why or why not? No, one would not expect lactate to accumulate in the muscle or blood of this patient because in the absence of glycogen breakdown to glucose‐1‐phosphate less glycolytic activity occurs to produce any excess pyruvate that would convert to lactate. 3. What fuels would you expect this patient to use during exercise? McArdle's patients primarily function with blood glucose metabolism, fat metabolism, and protein metabolism since carbohydrate metabolism cannot occur. 4. During what types of exercise would you expect this patient to experience the most problems? One would expect this patient to experience the most problems with high intensity aerobic activity because it utilizes almost 100% carbohydrates. When engaging in strenuous activity, it is advised that McArdle’s patients utilize a 6 second rule where s/he only performs the activity for 6 seconds prior to resting. This is to ensure that only the phosphogen energy system, or the ATP-CP system, is utilized. The phophogen energy system is anaerobic in addition to not producing lactic acid if oxygen is unavailable. 70/75 8