Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
RNA interference wikipedia , lookup
Genomic imprinting wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Public health genomics wikipedia , lookup
Genome evolution wikipedia , lookup
History of genetic engineering wikipedia , lookup
Ridge (biology) wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Pathogenomics wikipedia , lookup
Biology and consumer behaviour wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Quantitative trait locus wikipedia , lookup
Minimal genome wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Genome (book) wikipedia , lookup
Gene expression profiling wikipedia , lookup
Designer baby wikipedia , lookup
Microevolution wikipedia , lookup
Identifying Candidate Pathways to
Explain Phenotypes in
Genome-Wide Mutant Screens
Mark Craven
Department of Biostatistics & Medical Informatics
University of Wisconsin-Madison
U.S.A.
joint work with: Deborah Chasman,
Paul Ahlquist, Brandi Gancarz, Linhui Hao
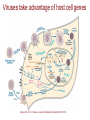
Viruses take advantage of host cell genes
Figure from: C. E. Samuel, Journal of Biological Chemistry 285, 2010.
Genome-wide mutant screens
mutant
phenotype
MED1
HOS1
LTP1
NOT5
LTP1
YPR071W
NOT5
SPE3
LTP1
MED1
HOS1
YPR071W
SPE3
MED1
HOS1
NOT5
SPE3
MED1
HOS1
YPR071W
SPE3
YPR071W
NOT5
LTP1
Example: determining which host genes affect viral replication
[Kushner et al., PNAS 2003; Gancarz et al., PLoS One 2011]
Genome-wide mutant screens
The output of such screens are sets of genes that either inhibit or
stimulate viral processes
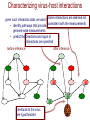
Characterizing virus-host interactions
given such interaction data, we want toSome interactions are deemed not
consistent with the measurements
• identify pathways that provide consistent explanations for the
genome-wide measurements
• predict the interfaces
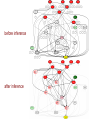
Directions to
and
thesigns
virusof
interactions are specified
before inference
after inference
Interfaces to the virus
are hypothesized
Integer programming approach
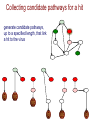
1. Collect candidate pathways for each “hit”
2. Use IP to identify a globally consistent subnetwork
Collecting candidate pathways for a hit
generate candidate pathways,
up to a specified length, that link
a hit to the virus
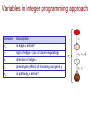
Variables in integer programming approach
x1
Variable
Description
xe
is edge e active?
se
sign of edge e (up- or down-regulating)
de
direction of edge e
tg
phenotype (effect) of knocking out gene g
σp
is pathway p active?
p
x2, s2, d2
tg
x 3, s3
Constraints in
integer programming approach
all significant measurements (hits) are
explained by at least one pathway
x1
n hits
p
0
p:n nodes( p )
p
x2, s2, d2
tg
x 3, s3
Constraints in
integer programming approach
all active pathways are consistent, with edges
directed toward the interface
p
p p 0 consistent ( p)
consistent ( p)
x1
x
e edges( p )
{i, j} nodes(e )
e
1 de dir( p,e) se t i t j
x2, s2, d2
tg
x 3, s3
Current objective function in
integer programming approach
minimize the number of interfaces
min
I p 0
n interf aces p:n nodes( p )
x1
p
x2, s2, d2
tg
x 3, s3
before inference
after inference
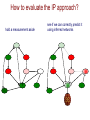
How to evaluate the IP approach?
hold a measurement aside
?
see if we can correctly predict it
using inferred networks
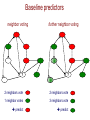
Baseline predictors
neighbor voting
?
further neighbor voting
?
2 neighbors vote
2 neighbors vote
1 neighbor votes
3 neighbors vote
predict
predict
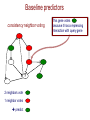
Baseline predictors
consistency neighbor voting
?
2 neighbors vote
1 neighbor votes
predict
This gene votes
because it has a repressing
interaction with query gene
Markov network approach
• variables are the same as in the IP
• potential functions represent
• the constraints
• uncertainty associated with specific interactions
• the preference for a small number of interfaces
• inference done using Gibbs sampling
Predictive Accuracy (BMV)
Predictive Accuracy (FHV)
Future work
• taking into account additional sources of information
• quantitative values from assays
• genetic interactions
• interactions automatically extracted from the scientific literature
• adapting approach to RNAi screens in mammalian cells
• more genes
• lower density of known interactions
• more uncertainty in measurements
• devising methods that use these models to determine which follow-up
experiments would be most informative