Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Molecular cloning wikipedia , lookup
Copy-number variation wikipedia , lookup
Real-time polymerase chain reaction wikipedia , lookup
Genetic engineering wikipedia , lookup
Transcriptional regulation wikipedia , lookup
Deoxyribozyme wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Two-hybrid screening wikipedia , lookup
Zinc finger nuclease wikipedia , lookup
Multilocus sequence typing wikipedia , lookup
Personalized medicine wikipedia , lookup
Molecular ecology wikipedia , lookup
Promoter (genetics) wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
DNA sequencing wikipedia , lookup
Silencer (genetics) wikipedia , lookup
Point mutation wikipedia , lookup
Bisulfite sequencing wikipedia , lookup
Exome sequencing wikipedia , lookup
Transposable element wikipedia , lookup
Community fingerprinting wikipedia , lookup
Non-coding DNA wikipedia , lookup
Endogenous retrovirus wikipedia , lookup
Whole genome sequencing wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Genomic library wikipedia , lookup
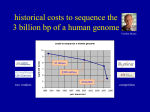
Last lecture summary Sequencing strategies • Hierarchical genome shotgun HGS – Human Genome Project • “map first, sequence second” • clone-by-clone … cloning is performed twice (BAC, plasmid) Sequencing strategies • Whole genome shotgun WGS – Celera • shotgun, no mapping • Coverage - the average number of reads representing a given nucleotide in the reconstructed sequence. HGS: 8, WGS: 20 Genome assembly • reads, contigs, scaffolds • base calling, sequence assembly, PHRED/PHRAP Human genome • 3 billions bps, ~20 000 – 25 000 genes • Only 1.1 – 1.4 % of the genome sequence codes for proteins. • State of completion: • best estimate – 92.3% is complete • problematic unfinished regions: centromeres, telomeres (both contain highly repetitive sequences), some unclosed gaps • It is likely that the centromeres and telomeres will remain unsequenced until new technology is developed • Genome is stored in databases • Primary database – Genebank (http://www.ncbi.nlm.nih.gov/sites/entrez?db=nucleotide) • Additional data and annotation, tools for visualizing and searching • UCSCS (http://genome.ucsc.edu) • Ensembl (http://www.ensembl.org) New stuff New generation sequencing (NGS) • The completion of human genome was just a start of modern DNA sequencing era – “high-throughput next generation sequencing” (NGS). • New approaches, reduce time and cost. • Holly Grail of sequencing – complete human genome below $ 1000. • Archon X Prize • http://genomics.xprize.org/ • $10 million prize is to be awarded to the private company that is able to sequence 100 human genomes within 10 days at cost of no more than $10 000 per genome 1st and 2nd generation of sequencers • 1st generation – ABI Prism 3700 (Sanger, fluorescence, 96 capillaries), used in HGP and in Celera • Sanger method overcomes NGS by the read length (600 bps) • 2nd generation - birth of HT-NGS in 2005. 454 Life Sciences developed GS 20 sequencer. Combines PCR with pyrosequencing. • Pyrosequencing – sequencing-by-synthesis • Relies on detection of pyrophosphate release on nucleotide incorporation rather than chain termination with ddNTs. • The release of pyrophosphate is detected by flash of light (chemiluminiscence). • Average read length: 400 bp • Roche GS-FLX 454 (successor of GS 20) used for J. Watson’s genome sequencing. 3rd generation • 2nd generation still uses PCR amplification which may introduce base sequence errors or favor certain sequences over others. • To overcome this, emerging 3rd generation of seqeuencers performs the single molecule sequencing (i.e. sequence is determined directly from one DNA molecule, no amplification or cloning). • Compared to 2nd generation these instruments offer higher throughput, longer read lengths (~1000 bps), higher accuracy, small amount of starting material, lower cost source: http://www.genome.gov/27541954 NHGRI Costs transition to 2nd generation $0.19 National Human Genome Research Institute (NHGRI) tracks the costs associated with sequencing. Which genomes were sequenced? • http://www.ncbi.nlm.nih.gov/sites/genome • GOLD – Genomes online database (http://www.genomesonline.org/) • information regarding complete and ongoing genome projects Important genomics projects • The analysis of personal genomes has demonstrated, how difficult is to draw medically or biologically relevant conclusions from individual sequences. • More genomes need to be sequenced to learn how genotype correlates with phenotype. • 1000 Genomes project (http://www.1000genomes.org/) started in 2009. Sequence the genomes of at least a 1000 people from around the world to create the detailed and medically useful picture of human genetic variation. • 2nd generation of sequencers is used in 1000 Genomes. • 10 000 Genomes will start soon. Important genomics projects • ENCODE project (ENCyclopedia Of DNA Elements, http://www.genome.gov/ENCODE/) • by NHGRI • identify all functional elements in the human genome sequence • Defined regions of the human genome corresponding to 30Mb (1%) have been selected. • These regions serve as the foundation on which to test and evaluate the effectiveness and efficiency of a diverse set of methods and technologies for finding various functional elements in human DNA. Rapid Evolution of Next Generation Sequencing Technologies 2000: Human genome working drafts Data unit of approximately 10x coverage of human 10 years and cost about $3 billion • 2008: Major genome centers can sequence the same number of base pairs every 4 days • • 1000 Genome project launched World-wide capacity dramatically increasing • 2009: Every 4 hours ($25,000) • 2010: Every 14 minutes ($5,000) • Illumina HiSeq2000 machine produces 200 gigabases per 8 day run cDNA 1. isolate mRNA from suitable cells 2. convert it to complementary DNA (cDNA) using the enzyme reverse transcriptase (+ DNA poymerase) • cDNA contains only expressed genes, no intergenic regions, no introns (just exons). • Because usually the desired gene sequences still represent only a tiny proportion of the total cDNA population, the cDNA fragments are amplified by cloning/PCR. • cDNA library – a library is defined simply as a collection of different DNA sequences that have been incorporated into a vector. ESTs • Expressed Sequence Tag • Their use was promoted by Craig Venter. At that time (1991) it was a revolutionary way for gene identification. • EST is a short subsequence (200-800 bps) of cDNA sequence. They are unedited, randomly selected singlepass sequence reads derived from cDNA libraries. • They can be generated either from 5’ or from 3’ end. mRNA cDNA 5’ ESTs 3’ ESTs ESTs • ESTs and cDNA sequences provide direct evidence for all the sampled transcripts and they are currently the most important resources for transcriptome exploration. • ESTs/cDNA sequences cover the genes expressed in the given tissue of the given organism under the given conditions. • housekeeping genes – gene products required by the cell under all growth conditions (genes for DNA polymerase, RNA polymerase, rRNA, tRNA, …) • tissue specific genes – different genes are expressed in the brain and in the liver, enzymes responding to a specific environmental condition such as DNA damage, … ESTs vs. whole genome • Whole genome sequencing is still impractical and expensive for organisms with large genome sizes. • Genome expansion, as a result of retrotransposon repeats, makes whole genome sequencing less attractive for plants such as maize. • Transposons - sequences of DNA that can move (transpose) themselves to new positions within the genome. • Retrotransposons – subclass of transposons, they can amplify themselves. Ubiquitous in eukaryotic organisms (45%-48% in mammals, 42% in human). Particularly abundant in plants (maize – 49-78%, wheat – 68%) • Genome expansion – increase in genome size, one of the elements of genome evolution EST properties • Individual raw EST has negligible biological information, it is just a very short copy of mRNA . • It is highly error prone, especially at the ends. The overall sequence quality is usually significantly better in the middle. Nagaraj SH, Gasser RB, Ranganathan S. A hitchhiker's guide to expressed sequence tag (EST) analysis. Brief Bioinform. 2007 8(1):6-21. PMID: 16772268. Problems in ESTs • Redundancy • Under-representation and over-representation of selected • • • • host transcripts (i.e. sequence bias) Base calling errors (as high as 5%) Contamination from vector sequences Repeats may pose problems Natural sequence variations (e.g. SNPs) – how to distinguish them and sequencing artifacts? ESTs on the web • Largest repository: dbEST (http://www.ncbi.nlm.nih.gov/dbEST/) • 1.7. 2011 – 69 992 536 ESTs from more than 1 000 organisms • UniGene (http://www.ncbi.nlm.nih.gov/unigene) stores unique genes and represents a nonredundant set of gene-oriented clusters generated from ESTs. EST analysis generic steps involved in EST analysis The aim of the analysis: augment weak signals, make consensus, when a multitude of ESTs are analysed reconstruct transcriptome of the organism. Nagaraj SH, Gasser RB, Ranganathan S. A hitchhiker's guide to expressed sequence tag (EST) analysis. Brief Bioinform. 2007 8(1):6-21. PMID: 16772268. EST preprocessing • Reduces the overall noise in EST data to improve the efficacy of subsequent analyses. • Remove vector contaminating fragments. • Compare ESTs with non-redundant vector databases (UniVec - EMVEC – http://www.ebi.ac.uk/Tools/sss/ncbiblast/vectors.html) http://www.ncbi.nlm.nih.gov/VecScreen/UniVec.html, • Repeats must be detected and masked using RepeatMasker (http://www.repeatmasker.org/). • Resources for EST pre-processing: page 12 in Nagaraj SH, Gasser RB, Ranganathan S. A hitchhiker's guide to expressed sequence tag (EST) analysis. Brief Bioinform. 2007 8(1):6-21. PMID: 16772268. EST clustering • Collect overlapping ESTs from the same transcript of a single gene into a unique cluster to reduce redundancy. • Clustering is based on the sequence similarity. • Different steps for EST clustering are described in detail in Ptitsyn A, Hide W. CLU: a new algorithm for EST clustering. BMC Bioinformatics. 2005; 6 Suppl 2:S3. PubMed PMID: 16026600 • The maximum informative consensus sequence is generated by ‘assembling’ these clusters, each of which could represent a putative gene. This step serves to elongate the sequence length by culling information from several short EST sequences simultaneously. • Sequence clustering and assembly: CAP3 Functional annotations • Database similarity searches (BLAST) are subsequently performed against relevant DNA databases and possible functionality is assigned for each query sequence if significant database matches are found. • Additionally, a consensus sequence can be conceptually translated to a putative peptide and then compared with protein sequence databases. Protein centric functional annotation, including domain and motif analysis, can be carried out using protein analysis tools. EST analysis pipelines • Large-scale sequencing projects (thousands of ESTs generated daily) – store, organize and annotate EST data in an automatic pipeline. • Database of raw chromatograms → clean, cluster, assemble, generate consensus, translate, assign putative function based on various DNA/protein similarity searches • examples: • TGI Clustering tools (TGICL) http://compbio.dfci.harvard.edu/tgi/software/ • PartiGene http://nebc.nerc.ac.uk/tools/other-tools/est Sequence Alignment What is sequence alignment ? CTTTTCAAGGCTTA GGCTTATTATTGC Fragments overlaps CTTTTCAAGGCTTA GGCTATTATTGC CTTTTCAAGGCTTA GGCT-ATTATTGC What is sequence alignment ? CCCCATGGTGGCGGCAGGTGACAG CATGGGGGAGGATGGGGACAGTCCGG TTACCCCATGGTGGCGGCTTGGGAAACTT TGGCGGCTCGGGACAGTCGCGCATAAT CCATGGTGGTGGCTGGGGATAGTA TGAGGCAGTCGCGCATAATTCCG “EST clustering” CCCCATGGTGGCGGCAGGTGACAG CATGGGGGAGGATGGGGACAGTCCGG TTACCCCATGGTGGCGGCTTGGGAAACTT TGGCGGCTCGGGACAGTCGCGCATAAT CCATGGTGGTGGCTGGGGATAGTA TGAGGCAGTCGCGCATAATTCCG TTACCCCATGGTGGCGGCTGGGGACAGTCGCGCATAATTCCG consensus Sequence alphabet side chain charge at physiological pH 7.4 Positively charged side chains Negatively charged side chains Polar uncharged side chains Special Hydrophobic side chains Name Arginine Histidine Lysine Aspartic Acid Glutamic Acid Serine Threonine Asparagine Glutamine Cysteine Selenocysteine Glycine Proline\ Alanine Leucine Isoleucine Methionine Phenylalanine Tryptophan Tyrosine Valine 3 letters Arg His Lys Asp Glu Ser Thr Asn Gln Cys Sec Gly Pro Ala Leu Ile Met Phe Trp Tyr Val 1 letter R H K D E S T N Q C U G P A L I M F W Y V Adenine A Thymine T Cytosine G Guanine C Sequence alignment • Procedure of comparing sequences • Point mutations – easy ACGTCTGATACGCCGTATAGTCTATCT ACGTCTGATTCGCCCTATCGTCTATCT gapless alignment • More difficult example ACGTCTGATACGCCGTATAGTCTATCT CTGATTCGCATCGTCTATCT • However, gaps can be inserted to get something like this insertion × deletion indel ACGTCTGATACGCCGTATAGTCTATCT ----CTGATTCGC---ATCGTCTATCT gapped alignment Why align sequences – continuation • The draft human genome is available • Automated gene finding is possible • Gene: AGTACGTATCGTATAGCGTAA • What does it do? • One approach: Is there a similar gene in another species? • Align sequences with known genes • Find the gene with the “best” match Flavors of sequence alignment pair-wise alignment × multiple sequence alignment Flavors of sequence alignment global alignment × local alignment global local align entire sequence stretches of sequence with the highest density of matches are aligned, generating islands of matches or subalignments in the aligned sequences Protein vs. DNA sequences • Given the choice of aligning DNA or protein, it is often more informative to compare protein sequences. • There are several reasons for this: • Many changes in DNA do not change the amino acid that is specified. • Many amino acids share related biophysical properties. Though these amino acids are not identical, they can be more easily substituted each with other. These relationships can be accounted for using scoring systems. • When is it appropriate to compare nucleic sequences? • confirming the identity of DNA sequence in database search, searching for polymorphisms, confirming identity of cloned cDNA Evolution of sequences • The sequences are the products of molecular evolution. • When sequences share a common ancestor, they tend to exhibit similarity in their sequences, structures and biological functions. DNA1 DNA2 Protein1 Protein2 Sequence similarity Similar 3D structure Similar function Similar sequences produce similar proteins However, this statement is not a rule. See Gerlt JA, Babbitt PC. Can sequence determine function? Genome Biol. 2000;1(5) PMID: 11178260