Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Transposable element wikipedia , lookup
Protein moonlighting wikipedia , lookup
Gene expression profiling wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Point mutation wikipedia , lookup
Minimal genome wikipedia , lookup
Whole genome sequencing wikipedia , lookup
Non-coding DNA wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Pathogenomics wikipedia , lookup
Genomic library wikipedia , lookup
Metagenomics wikipedia , lookup
Helitron (biology) wikipedia , lookup
Human genome wikipedia , lookup
Multiple sequence alignment wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Human Genome Project wikipedia , lookup
Sequence alignment wikipedia , lookup
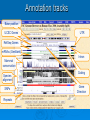
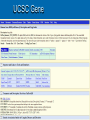
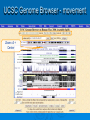
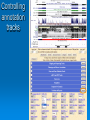
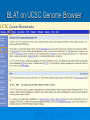
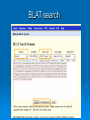
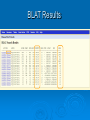
Psi-BLAST, Prosite, UCSC Genome Browser Lecture 3 Searching for remote homologs Sometimes BLAST isn’t enough Large protein family, and BLAST only finds close members. We want more distant members PSI-BLAST Position Specific Iterative BLAST Regular blast Construct profile from blast results Blast profile search Final results Consensus, Pattern, PSSM 1 Seq1 Seq2 Seq3 2 3 4 5 6 A T C T T G A A C T T G A A C T T C Consensus: Pattern: Profile = PSSM: the most frequent character in the column is chosen represents the alignment as a regular expression Position Specific Score Matrix Pos 1 2 3 4 5 6 A 1 .67 0 0 0 0 C 0 0 1 0 0 .33 G 0 0 0 0 0 .67 T 0 .33 0 1 1 0 Nuc AAC T T G A-[TA]-C-T-T-[GC] Pos Nuc A C G T 1 1 0 0 0 2 .67 .33 0 0 3 0 1 0 0 4 0 1 0 0 5 .25 .25 .25 .25 6 .33 0 .33 .33 S(AACCAA)=1*0.67*1*1*.25*.33 S(GACCAA)=0 Sequences with higher scores -> higher chance of being related to the PSSM PSI-BLAST Position Specific Iterative BLAST Regular blast Construct profile from blast results Blast profile search Final results BLAST – PSI-Blast PSI-Blast - results PSI-BLAST PSI-BLAST looks for seq’s that are close to the query, and learns from them to extend the circle of friends Disadvantage: if we obtained a WRONG hit, we will get to unrelated sequences (contamination). This gets worse and worse each iteration Advantage: PSI-BLAST Which of the following is/are correct? 1. PSI-BLAST is expected to give more hits than BLAST 2. PSI-BLAST is an iterative search method 3. PSI-BLAST is faster than BLAST 4. Each iteration of PSI-BLAST can only improve the results of the previous iteration Turning information into knowledge The outcome of a sequencing project are masses of raw data The challenge is to turn these raw data into biological knowledge A valuable tool for this challenge is an automated diagnostic pipe through which newly determined sequences can be streamlined From sequence to function Nature tends to innovate rather than invent Proteins are composed of functional elements: domains and motifs Domains are structural units that carry out a certain function. They are shared between different proteins Motifs are shorter and are usually critical for the biological activity http://www.expasy.ch/prosite Prosite From analyzing conserved regions in protein sequences it is possible to derive signatures of motifs and domains Prosite consists of annotated sites/motifs/signatures/fingerprints Given an uncharacterized translated protein sequence, prosite tries to predict which motifs and domains make up the protein and thus identify the family to which it belongs Prosite Prosite represents entries with patterns or profiles ATCTTG AA C T T G AA C T T C profile pattern A-[TA]-C-T-T-[GC] 1 2 3 4 5 6 A 1 0.67 0 0 0 0 T 0 0.33 0 1 1 0 C 0 0 1 0 0 0.33 G 0 0 0 0 0 0.67 Profiles are used in prosite when the motif is relatively divergent, and is difficult to represent as a pattern Profiles also characterize domains over their entire length, not just the motif Prosite sequence query Patterns with a high probability of occurrence Entries describing commonly found posttranslational modifications or compositionally biased regions Found in the majority of known protein sequences High probability of occurrence Prosite filters them by default Scanning Prosite Query: sequence Result: all patterns found in the sequence Query: pattern Result: all sequences which adhere to this pattern Prosite pattern query UCSC Genome Browser UCSC Genome Browser Gateway Reset all settings of previous uses UCSC Genome Browser Gateway Results Annotation tracks Base position UCSC Genes UTR RefSeq Genes mRNAs (GenBank) Intron Mammal conservation Species alignment SNPs Repeats Coding Gene Direction UCSC Gene UCSC Genome Browser - movement Zoom x3 + Center Controlling annotation tracks Sickle-cell anemia distr. Malaria distr. BLAT BLAT = Blast-Like Alignment Tool BLAT is designed to find similarity of >95% on DNA, >80% for protein Rapid search by indexing entire genome Good for: 1. Finding genomic coordinates of cDNA 2. Determining exons/introns 3. Finding human (or chimp, dog, cow…) homologs of another vertebrate sequence BLAT on UCSC Genome Browser BLAT search BLAT Results BLAT Results query Match hit Non-Match (mismatch/indel) Indel boundaries