Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Silencer (genetics) wikipedia , lookup
Biochemistry wikipedia , lookup
Gene expression wikipedia , lookup
Magnesium transporter wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Protein (nutrient) wikipedia , lookup
Metalloprotein wikipedia , lookup
Circular dichroism wikipedia , lookup
Interactome wikipedia , lookup
List of types of proteins wikipedia , lookup
Protein moonlighting wikipedia , lookup
Protein folding wikipedia , lookup
Western blot wikipedia , lookup
Protein domain wikipedia , lookup
Homology modeling wikipedia , lookup
Protein adsorption wikipedia , lookup
Protein–protein interaction wikipedia , lookup
Intrinsically disordered proteins wikipedia , lookup
X-ray crystallography wikipedia , lookup
Protein structure prediction wikipedia , lookup
Nuclear magnetic resonance spectroscopy of proteins wikipedia , lookup
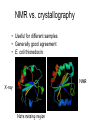
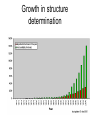
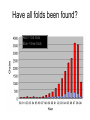
X-ray crystallography – an overview (based on Bernie Brown’s talk, Dept. of Chemistry, WFU) • Protein is crystallized (sometimes low-gravity atmosphere is helpful e.g. NASA) • X-Rays are scattered by electrons in molecule • Diffraction produces a pattern of spots on a film that must be mathematically deconstructed • Result is electron density (contour map) – need to know protein sequence and match it to density • Hydrogen atoms not typically visible (except at very high resolution) X-ray Crystallography – in a nutshell REFLECTIONS Bragg’s law h k l 0 0 0 0 0 . . . 0 0 0 0 0 I σ(I) 2 3523.1 3 -1.4 4 306.5 5 -0.1 6 10378.4 91.3 2.8 9.6 4.7 179.8 Fourier transform ? Phase Problem ? MIR MAD MR Electron density: r(x y z) = 1/V SSS |F(h k l)| exp[–2pi (hx + hy + lz) + ia(h k l)] Crystal formation • Start with supersaturated solution of protein • Slowly eliminate water from the protein • Add molecules that compete with the protein for water (3 types: salts, organic solvents, PEGs) • Trial and error • Most crystals ~50% solvent • Crystals may be very fragile Visible light vs. X-rays Why don’t we just use a microscope to look at proteins? • Size of objects imaged limited by wavelength. Resolution ~ l/2 – Visible light – 4000-7000 Å (400-700 nm) – X-rays – 0.7-1.5 Å (0.07-0.15 nm) • It is very difficult to focus X-rays (Fresnel lenses) • Getting around the problem – Defined beam – Regular structure of object (crystal) • Result – diffraction pattern (not a focused image). Diffraction pattern – lots of spots Bragg’s Law: 2d sinq = nl X-ray beam crystal ~1015 molecules/crystal Diffraction pattern is amplified Film/Image plate/CCD camera End result – really! Fourier transform of diffraction spots electron density fit a.a. sequence DNA pieces Protein (Dimer of dimers) Interference of waves • In crystallography, get intensity information only, not phase information • Need to deconvolute and obtain phase information: • THE PHASE PROBLEM How to get from spots to structure? • Fourier synthesis • Getting around phase problem – Trial and error – Previous structures – Heavy atom replacement – make a landmark – Ex: Selenomethionine • Plenty of computer algorithms now Electron density with incorrect phases • Red is true structure The effect of resolution More extensive diffraction pattern gives more structural information = higher resolution • 6.0-4.5 Å – secondary structure elements • 3.0 Å – trace polypeptide chain • 2.0 Å – side chain, bound water identification • 1.8 Å – alternate side chain orientations • 1.2 Å – hydrogen atoms With computational tools, spots become density Flexible regions give smeared density, often 2-3 conformations visible, more than that invisible Density becomes structure Need to know protein sequence to trace backbone Co-crystal structures • Because of relatively high solvent content, can often “soak in” substrate • Then can solve structure of protein with substrate bound • If crystal cracks, good sign that substrate binding or enzyme catalysis results in conformational change in protein • No longer has same crystal arrangement NMR vs. crystallography • Useful for different samples • Generally good agreement • E. coli thioredoxin: NMR X-ray Note missing region Known protein structures • ~17,000 protein structures since 1958 • Common depository of x,y,z coordinates: Protein data bank (http://www.rcsb.org) • Coordinates can be extracted and viewed • Comparisons of structures allows identification of structural motifs • Proteins with similar functions and sequences = homologs Growth in structure determination Function from structure • Might identify a pocket lined with negatively-charged residues • Or positively charged surface – possibly for binding a negatively charged nucleic acid • Rossmann fold – binds nucleotides • Zinc finger – may bind DNA Domain organization • Large proteins have polypeptide regions that fold in isolation • May have distinct functional roles – Example: glyceraldehyde-3phosphate dehydrogenase Protein families • Similar function and overall structure • But amino acid sequence may or may not be highly conserved • Limited number of protein domains • Homologs versus structural motifs SCOP Classification Statistics Structural Classification of Proteins 18946 PDB Entries, 49497 Domains (1 March 2002) (excluding nucleic acids and theoretical models) Class Folds All a All b Alpha & beta (a/b) Alpha & beta (a+b) Multi-domain proteins Superfamilies Families Membrane /cell-surface proteins 171 119 117 224 39 34 286 234 192 330 39 64 457 418 501 532 50 128 Small proteins Total 61 765 87 1232 135 2164 http://scop.berkeley.edu/ or http://scop.mrc-lmb.cam.ac.uk/scop/ Have all folds been found? Red = Old folds Blue = New folds