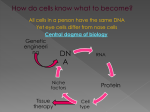
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Genetic code wikipedia , lookup
Biochemistry wikipedia , lookup
Transcriptional regulation wikipedia , lookup
Promoter (genetics) wikipedia , lookup
Endogenous retrovirus wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
Community fingerprinting wikipedia , lookup
Gene nomenclature wikipedia , lookup
Point mutation wikipedia , lookup
Magnesium transporter wikipedia , lookup
Gene regulatory network wikipedia , lookup
Western blot wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
Structural alignment wikipedia , lookup
Gene expression wikipedia , lookup
Metalloprotein wikipedia , lookup
Expression vector wikipedia , lookup
Silencer (genetics) wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Nuclear magnetic resonance spectroscopy of proteins wikipedia , lookup
Proteolysis wikipedia , lookup
Interactome wikipedia , lookup
Proteiinianalyysi 5 Rakenteen ennustaminen Funktion ennustaminen http://www.bioinfo.biocenter.helsinki.fi/downlo ads/teaching/spring2005/proteiinianalyysi/ Sekvenssistä rakenteeseen • komparatiivinen mallitus • 1-ulotteinen tilan (luokan) ennustaminen sekvenssistä • 3-ulotteisen rakenteen tunnistaminen annetusta kirjastosta (fold recognition) • 3-ulotteisen rakenteen ennustaminen ab initio – ROSETTA The “Folding Problem” Two parts: (1) The “Search Problem” Is the true structure one of my 2 million guesses? Fragment assembly (2) The “Discrimination Problem” If it’s one of these 2 million, which one is it? Empirical pseudopotential Rosetta (1) A stone with three ancient languages on it. (2) A program (David Baker) that simulates the folding of a protein, using statistical energies and moves. Fold prediction – Rosetta method • Knowledge based scoring function Bayes' law: P(structure) * P(sequence|structure) P(structure|sequence) = P(sequence) P(sequence|structure) = f(residue contacts in native structures) sequence consistent local structure protein-like structures near-native structures P(structure) = probability of a protein-like structure (no clashes, globular shape) Simons et al. (1997) Collection of putative backbone conformations Protein sequence Library of small segments ... ... For each window of 9 residues: lookup 25 closest (sequence) neighbours in library ... sequences structures Simons et al. (1997) Intermediates are not observed, but Folding is 2-state Unfolded Folded Nucleation sites something happens first... Early folding events might be recorded in the database Non-homologous proteins Short, recurrent sequence patterns could be folding Initiation sites recurrent part HDFPIEGGDSPMQTIFFWSNANAKLSHGY CPYDNIWMQTIFFNQSAAVYSVLHLIFLT IDMNPQGSIEMQTIFFGYAESA ELSPVVNFLEEMQTIFFISGFTQTANSD INWGSMQTIFFEEWQLMNVMDKIPS IFNESKKKGIAMQTIFFILSGR PPPMQTIFFVIVNYNESKHALWCSVD PWMWNLMQTIFFISQQVIEIPS MQTIFFVFSHDEQMKLKGLKGA Nature has selected for these patterns because they speed folding. I-sites motifs diverging type-2 turn Serine hairpin Proline helix C-cap Backbone angles: y=green, f=red Amino acids arranged from nonpolar to polar alpha-alpha corner Type-I hairpin Frayed helix glycine helix N-cap Rosetta Fragment insertion Monte Carlo backbone torsion angles moveset accept or reject Choose fragment from moveset change backbone angles Convert angles to 3D coordinates Energy function Rosetta Backbone angles are restrained in I-sites regions regions of highconfidence I-sites prediction moveset backbone torsion angles Fragments that deviate from the paradigm (>90° in f or y) are removed from the moveset. Generally, about one-third of the sequence has an I-sites prediction with confidence > 0.75, and is restrained. Rosetta Sequence dependent features Rosetta Sequence-independent features Current structure vector representation Probabilities from the database The energy score for a contact between secondary structures is summed using database statistics. MC-SA optimization • for each random position – pick a random neighbour – replace backbone conformation – calculate probability of new structure • MC: Monte-Carlo – accept up-hill moves with a certain probability that depends on temperature • SA: simulated annealing – Gradual cooling of temperature: first allow many changes, later fewer changes Simons et al. (1997) Results • Small molecules: ok • Proteins with mostly α-helices: ok • Proteins with mostly β-sheets: not so ok Simons et al. (1997) Rosetta What needs to be fixed? Turns 8% of the residues in the targets have f > 0. 44% of these are at Glycine residues. 7% of the residues in the predictions have f > 0. but only 16% of these are at Glycines. Contact order N 1 CO Sij LN True structure: 0.252 Predictions: 0.119 Prediction algorithms have underlying principles Darwin = protein evolution. Principle: Proteins that evolved from common ancestor have the same fold. Boltzmann = protein folding Principle: Proteins search conformational space, minimizing the free energy (empirical pseudo-potential) Geenin funktion määrittäminen • fenotyyppi • biokemiallinen aktiivisuus (in vitro) • ilmentyminen • GO, Gene Ontology – molekulaarinen funktio – biologinen prosessi – solunsisäinen lokalisaatio Homologia sama funktio? Paralogia: geenien kahdentumisen tulos Vaihtoehtoinen silmukointi: yksi geeni, monta proteiinia Pleiotropia: yksi geeni, monta funktiota Redundanssi: yksi funktio, monta geeniä Heteromeria: kompleksien muodostus “Crosstalk”: signalointireitit vaikuttavat toisiinsa Protein functional shifts are common • COG0044 – Dihydroorotase • CAD (fusion protein) – Dihydropyriminidase – D-hydantoinase – Allantoinase – Rudimentary protein (involved in developmental programs) COG0044 functions Urease superfamily functions Fast evolution ~ functional shift rat lung isoform rat liver isoform, functional shift CYP2 family (cytochrome P450) “Druggable genome” • Property filters – Likelihood of functional shift – Degree and nature of paralogy – Factors reflecting pleiotropy • • • • Size Breadth of expression Interaction potential Evolutionary rates Funktion siirto • Nearest neighbour (lähin homologi) – esim. Blast-haku – Fylogeneettinen lähin naapuri • Post-genomiset menetelmät – riippumattomia homologiasta – Proteiini-proteiini-interaktioiden vertailu • Guilt By Association • Hahmontunnistus Funktion siirto • Hypoteettinen sekvenssi funktio? – Karakterisoitu homologi • Blast / PSI-Blast – Fylogenia! • evoluutionopeus riippuu perheestä • monen sekvenssin linjaus • Virheelliset funktion määritykset kertautuvat tietokannoissa! – Väärä funktio • liittyy domeeniin, jota ei esiinny hakusekvenssissä – Väärä homologiapäätelmä – Liian yksityiskohtainen funktion kuvaus • funktion muuttuminen evoluutiossa • biokemiallinen vs. fysiologinen funktio – esim. eukaryoottispesifiset funktiot eivät voi esiintyä bakteerissa – Sekvenssilinjaus • funktionaalisten aminohappojen säilyminen • esimerkki: – atratsiiniklorohydrolaasi vs. melamiinideaminaasi: 4 mutaatiota (98 % identtisyys) – Esim. GO liputtaa funktion määrityksen lähteen Guilt by association • Prediction of subcellular localization based on classification of neighbours Non-homology protein identification using network context Query pattern Interactome Ref: Lappe M, Park J, Niggemann O, Holm L (2001) Bioinformatics Suppl 1, S149-S156 Natural selection • Functional coupling leads to correlations – E.g. co-occurrence of sets of genes in species • Residues required for molecular function – Functional conservation above general sequence divergence of a family Pancreatic trypsin inhibitor (2ptc) Approaches • Evolutionary Trace – Lichtarge et al. 1996 • Sequence Space – Casari et al. 1995 • Ortholog / paralog discriminants – Mirny & Gelfand 2003 Evolutionary Trace • The branchpoints separating subclades of a phylogenetic tree can specify molecular speciation events, and hence evolutionary selection of amino acids • Map trace residues to 3D structures Evaluation of Evolutionary Trace • Trace residues determined at many ranks – Trace residue sets are nested • Test of significance of trace residue at any rank – Overlap with otherwise defined functional sites • Bound ligands in 3D structures (~20 residues) • Annotated sites (~4 residues) ET assessment • Detects 3D clusters • Manual filtering and pruning of the data – Decide which subclades of the protein family to use in analysis – Exclude fragments – Original method was based on strict invariance within subclade • Automatic implementations – But manually optimized traces score higher Sequence Space • Aligned protein sequences represented as vectors in a high-dimensional space – Each amino acid type at each column of the MSA is a unique point in Sequence Space • Dimension reduction by Principal Components Analysis • Cluster proteins – Based on their sequence identity • Map residues in the same space – Direction points to association with protein group A 3D object PCA projection of the 3D object New axes are linear combination of original axes Coding of amino acids Sequence vector representation Interpretation 1st axis represents the whole family 2nd, 3rd , …, 6th axes represent subclassifications Subfamily-specific residues are found at the tips of a polygon Common residues shared by several subfamilies are found along the edges of a polygon Many unspecific residues at origin Protein clustering Residue clustering Selection of residues & proteins Ortologit ja paralogit Malliorganismien käyttö: identtinen fysiologia? Summary • Functional groupings of proteins – Phylogenetic lineage • Orthologs / paralogs – Clustering by general sequence similarity • Residues associated with above groupings – Intra-group conservation – Inter-group variation – Neutral residues behave randomly Function = interactions • Protein-protein interactions • Co-evolution of interacting proteins • Comparative genomics Experimental methods • Y2H = yeast-two-hybrid – – – – Ex vivo, binary interactions Interaction must occur in the nucleus Autoactivation (5-10 % of random ORFs) Posttranslational modifications • AP/MS = affinity purification / Mass Spectrometry – Purified complexes • PChips = protein microarrays – In vitro – Covalent attachment to solid support – Screening with fluorescently labelled probes (e.g. proteins or lipids) Small part of an interaction network NewScientist, 13. April 2002, David Cohen about the work by Barabasi, Albert et al. Interaktioiden ennustaminen • ko-evoluutio • genomien vertailu – geenien järjestys kromosomissa – fylogeneettiset profiilit – geenifuusio Ko-evoluutio monen sekvenssin linjaus, etsi korreloivat mutaatiot • proteiinit, joilla on paljon interaktioita, muuttuvat hitaammin • kaksi fylogeniapuuta, etsi parit • Comparative genomics • Correlated genomic context between orthologous genes reveal functional couplings – Conserved gene order (conserved synteny) – Coupled gene loss / preservation (phylogenetic profiles) – Gene fusion events Conserved synteny • Chromosomal rearrangements randomize gene order over the course of evolution • Groups of genes that have a similar biological function tend to remain localized in a group or cluster • Bacterial operons allow coordinated regulation of gene expression from a common promoter • Eukaryotic clusters observed, too Phylogenetic profiling p1 p4 p5 p1 p2 p3 p5 p6 p8 yeast H. influenzae p2 p3 p4 p5 p7 E. coli ye P1 1 P2 1 P3 1 P4 0 P5 1 P6 1 P7 0 P8 1 hi 1 0 0 1 1 0 0 0 ec 0 1 1 1 1 0 1 0 ye P7 0 P4 0 P6 1 P8 1 P2 1 P3 1 P1 1 P5 1 hi 0 1 0 0 0 0 1 1 ec 1 1 0 0 1 1 0 1 Observations - phyloprofiles • Bit-vectors sensitive to noise in gene status assignment • Specific patterns generated mainly from bacterial gene loss / horizontal transfer • Eukaryotic species have larger genomes and large numbers of eukaryote-specific protein families Gene fusion Domain swapping Some details • 6,809 interactions predicted for E. coli based on gene fusions – 321 (~5 %) overlap with predictios by phylogenetic profile method – Eight times more than random • Promiscuous modules (SH2, SH3, etc.) – 5 % of domains made more than 25 links to other proteins – Fusions counted within remaining set of 95 % Observations – gene fusion • Marcotte et al. (Science 285:751-753, 1999) predicted novel interactions for 50 % of yeast proteins using gene fusion information in any homologous proteins • Enright et al. (Nature 402:86-90, 1999) considered orthologs with higher signal-tonoise ratio but only 7 % coverage Integrated predictions • Predictions by conserved synteny, phylogenetic profiles and gene fusion are largely additive – small overlap • Combined score – Calibrated against same / different KEGG map • STRING server – Predictions for about 50 % of genes from complete genomes – http://www.bork.embl-heidelberg.de/STRING/ Functional association maps • Noisy • Different types of interaction – Physical interaction (complex formation) – Transient interactions • Dependent on post-translational modification state, e.g. phosphorylation – Functional linkage • Successive steps of a metabolic pathway • Involvement in related biological processes Tentti • Tentti 28.4. • Uusinta 3.5. yleinen tenttipäivä • Tenttiin tulee – Päättelytehtäviä – Esseekysymyksiä