Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Haemodynamic response wikipedia , lookup
Neuroplasticity wikipedia , lookup
Activity-dependent plasticity wikipedia , lookup
End-plate potential wikipedia , lookup
Embodied language processing wikipedia , lookup
Synaptogenesis wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Central pattern generator wikipedia , lookup
Electromyography wikipedia , lookup
Microneurography wikipedia , lookup
Muscle memory wikipedia , lookup
Neuromuscular junction wikipedia , lookup
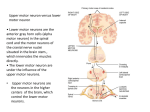
Module 3 The integration of postural control and selective movement for functional activities, Part B (Formerly Part B ‘management of the patient with established movement dysfunction’) This module will further develop movement analysis and therapeutic handling skills to enhance selective movement for locomotion, and functional reach. The aims of the module are: • • • • To discuss the upper motor neurone syndrome and its consequences for the patient with established movement dysfunction Practical sessions with an emphasis on distal key points Patient assessment and treatment To link evidence base and clinical practice The pathophysiology of the upper motor neurone (UMN) syndrome is complex (Sheean 2001) and our understanding of the underlying mechanisms, the interaction of neural and biomechanical changes and the significance of the sequalae continues to evolve. Gracies (2005) describes how paresis and/or muscle weakness that occurs as the initial consequence of neurological insult imposes a level of immobility on the individual. Neuroplastic adaptation follows leading to the development of the positive features of the UMN syndrome. The biomechanical consequences of initial immobility may be exacerbated by muscle overactivity which in turn increases the development of contracture. Muscle tone is the sensation of resistance that is encountered as a joint is passively moved through a range of movement. When an individual is at rest the resistance felt is due to a combination of the physical inertia of the limb and the viscoelastic properties of the muscles and connective tissues. Tension within the muscle due to reflex contraction (tonic stretch reflexes) contributes to the resistance only during movement in the neurologically intact adult. (Morris 2002). To understand the features of the UMN syndrome a review of spinal reflex circuitry is necessary. There are two types of spinal segmental reflexes: afferent disinhibited spinal segmental reflexes – proprioceptive, cutaneous and nociceptive information from the periphery; efferent – tonic supraspinal drive (related to cortical / brainstem regions of CNS). The muscle spindle located within the belly of the muscle has its own efferent supply via the gamma system to maintain tension in spindle fibres as the extrafusal muscle fibres © BBTA 2016 1 contract. The muscle spindle signals change in length of the muscle. The action potential generated is transferred along the large 1a afferent via the dorsal route ganglion to the alpha motor neuron within the spinal cord where it: mediates a stretch reflex through local spinal circuitry; ascends to mediate transcortical reflex like activity. Within the spinal cord 1a fibres from the muscle spindle make monosynaptic excitatory connections to alpha motor neurons of the same muscle and that of its synergists. A further connection to the 1a inhibitory interneurone inhibits activity in the antagonist. This forms the basis of reciprocal inhibition, i.e. excitation of agonist/inhibition of antagonist. As the 1a inhibitory interneurone receives both excitatory and inhibitory inputs from the descending pathways, lack of supraspinal control can decrease the level of reciprocal inhibition leading to more co-contraction. This combination of influences is described as reciprocal innervation. Reciprocal innervation is important not only during reflex activity but also during voluntary movement. The degree of stiffness around a joint can therefore be modified according to task requirements. A collateral from the motor neurone axon excites the Renshaw cell, another inhibitory interneurone, it then inhibits its own motor neurone and other neighboring populations of motor neurons. This creates a negative feedback system, which helps to stabilize the firing rate of the motor neurone. The motor neurones and inhibitory interneurons receive multiple inputs from the descending supraspinal pathways, from afferent input from the periphery and within the body allowing further modulation of output. The afferent from another sensory receptor must also be considered. The golgi tendon organ (GTO) located at the musculotendinous junction responds to changes in tension within the muscle. As the muscle contracts tension increases compressing the GTO. The action potential travels along the 1b afferent via the dorsal root ganglion to the 1b inhibitory interneurone, which then inhibits the activity of the alpha motor neurone also known as autogenic inhibition. As described above convergent input from within the body, the periphery and via descending pathways from supraspinal areas acting upon the !b inhibitory interneurone modulates output. A further consideration of the 1b system is that state dependent reflex reversal means that during locomotion or in a loaded posture the 1b system results in excitation rather than inhibition. Most reflex pathways are disynaptic or polysynaptic, with one or more interneurons between sensory and motor neurons. Supraspinal centres are able to co-ordinate muscle activity around a joint. Positive features of the UMN may be considered as an aberrant sensorimotor behaviour due to either the primary disruption of descending control or the resultant functional and structural reorganisation within the nervous system. Movement disorders as part of the UMN syndrome vary in different conditions e.g. stroke, MS, SCI. Lesion location determines the presentation of features of the UMN syndrome. An understanding of the descending control is therefore essential to appreciate the significance of lesion location. © BBTA 2016 2 A balance of modulation between the excitatory medial reticulospinal and vestibulospinal and inhibitory lateral reticulospinal systems determines the overall motor behaviour at a movement level. This can then be influenced and further modulated by inputs from other descending systems. Disruption to the descending systems and / or pathways therefore results in alterations to the levels of excitation and inhibition and resultant negative and positive features seen within the individuals presentation following brain or spinal cord injury or disease. Following a cortical lesion e.g. stroke the descending influence from the cortex on to the brainstem and spinal cord is interrupted reducing the (inhibitory) influence to the alpha motoneurone. A discrete lesion interrupting the lateral corticospinal pathway will result in distal weakness. A lesion within the brain stem influencing the ventromedial reticular formation will result in a further loss of inhibitory influences and the balance of modulation is tipped towards excitation. The resulting presentation is characterised by more positive features of the UMN syndrome. A lesion interrupting the dorsal (lateral) reticulospinal pathway within the spinal cord results in a similar loss of inhibition. A complete lesion of the brainstem/spinal cord interrupting ALL of the descending systems results in a loss of all inhibitory and excitatory influences. Motor behaviour is therefore at the behest of the peripheral input on the spinal reflex circuitry. Voluntary movement control is no longer a possibility following this type of lesion. The initial presentation following an UMN lesion is a result of the disruption of descending spinal control and includes: weakness, disinhibition and hyperexcitation. The clinical presentation (positive and negative features develops over time following structural and functional reorganisation of the nervous system as a result of processes such as: denervation super-sensitivity, collateral sprouting and unmasking of latent synapses. Clinical features of the UMN syndrome are described as either negative (loss of features) or positive (new features). Positive features are characterised by muscle over-activity and/or co-contraction. Negative features Weakness Loss of dexterity Fatiguability Acute hypotonia Loss of postural responses Positive features Spasticity Hyperreflexia Positive Babinski response Dysynergic patterns of co contraction Spasms Positive feature can further be described as those occurring at rest or during movement. Spasticity is only one feature of the UMN syndrome, but is a term that is frequently used to describe some or all positive features of the UMN syndrome. There is no universal definition of spasticity nor agreement about how it should be measured (Malhotra et al 2009). The definition of spasticity from Lance (1980) is perhaps the most widely used and © BBTA 2016 3 states that spasticity is a motor disorder characterised by a velocity – dependent increase in tonic stretch reflexes (muscle tone) with exaggerated tendon jerks (phasic stretch reflex), resulting from hyper excitability of the stretch reflex, as one component of the upper motor neurone syndrome. This definition was considered by many to be narrow and outdated. Three areas were identified as requiring modification within any definition of spastcity: 1. Velocity dependent changes in limb stiffness during passive movement are not solely due to neural changes but are contributed to by the normal viscoelastic properties of soft tissues 2. In addition to hyperexcitable stretch reflexes, activity in other pathways (afferent, supraspinal and changes in alpha motoneurone) is also important in the development of spasticity 3. Spasticity cannot be exclusively considered a ‘motor disorder’ as afferent activity (cutaneous and proprioceptive) is also involved The European Assembly for Spasticity Measurement (EU SPASM) in 2005 published a new definition of spasticity as: “disordered sensory-motor control, resulting from an upper motor neuron lesion, presenting as intermittent or sustained involuntary activation of muscles”. This new definition is considered to be broad enough to include all the positive features of the UMN syndrome and yet still exclude secondary biomechanical changes which although they contribute to stiffness and resistance to movement are not a primary feature of the UMN syndrome. There is evidence to suggest that hyper-reflexia results from a failure to modulate tonic stretch reflexes, rather than only hyper-excitability of the reflex. Singer et al 2001 describe spasticity as “a motor disorder in which failure to actively inhibit velocity sensitive stretch reflexes can lead to exaggerated muscle resistance to both externally and self-imposed muscle stretch and consequently, to impaired voluntary movement.” The physiological mechanisms underlying changes in the excitability of the reflex circuitry include: • Increased fusimotor drive • Alpha motoneurone hyperexcitability • An increase in the gain of the spinal reflex circuitry • A lowering of the threshold of the spinal circuitry It is important to remember that the spastic movement disorder and presenting features are not only different in different conditions e.g. stroke and SCI but also vary over time (Bennett 2008). In the clinical setting muscle tone is assessed by moving the limb and feeling the resistance as the limb is moved passively. The resistance that is felt when limb is moved at rest is a combination of the physical inertia of the extremity and the inherent viscoelastic properties of the muscle and connective tissue. The tension set up in the muscle by reflex contraction caused by muscle stretch contributes to the resistance only during movement. © BBTA 2016 4 Following an upper motoneurone lesion patients’ will try to function within their environment as best they can. For the majority of patients this will involve coping with postural instability and displacement, against a background of abnormal postural alignment and low tone / weakness In some patients this may result in an associated reaction. Associated reactions frequently occur in response to a loss of anticipatory postural adjustments during perturbations. Perturbations may occur as a result of activities such as sneezing, laughing, or effortful movement. Should the associated reaction be repeated frequently biomechanical changes to muscle and soft tissue length may occur. The limited or altered movement that occurs as a result of the neural consequences of the UMN syndrome also results in changes to both the myogenic and arthrogenic structures within the body. Changes within the muscles include a loss of sarcomeres in muscles held in a shortened position, altered protein synthesis with a proportional increase in the amount of collagen within muscle. Muscle fibre type has been shown to alter in response to the changed demand and cross bridge formation and release has also been implicated in changes to muscle compliance. Changes in the collagenous structures around and within joints include reductions in lubrication and alignment of collagen fibres and results in intra-articular adhesions (Singer et al 2001). The relative contribution of the neural and non-neural changes varies between individuals and over time. The contribution of non-neural changes to altered alignment of soft tissues, joints and limbs and the contribution to the movement disorders seen post UMN damage can be significant. In the assessment of a patient it is essential to consider • • • • • • • Alignment Muscle length Postural control / balance Activity Compensation Function Proprioceptive input Consideration should be given to the environment in which the patient is functioning, the postures the patient adopts over the 24 hours, the assistance that the patient receives. It is also essential to be aware of any pharmacological management of increased tone as well as any strategies such as splinting being used to manage biomechanical changes. In the treatment of the patient careful emphasis on optimal alignment of both muscles and joints within the limbs and trunk will ensure optimal potential for recovery of postural control, selective movement and function. Reading Ada L, O’Dwyer N, O’Neill E. (2006) Relation between spasticity, weakness and contracture of the elbow flexors and upper limb activity after stroke: an observational study Disability and Rehabilitation 28: 891-897. © BBTA 2016 5 Adams MM, Hicks AL. (2005) Spasticity after spinal cord injury. Spinal Cord 43: 577-586. Azari N, Seitz R. (2000) ‘Brain plasticity and recovery from stroke’. American Scientist 88:426-31 Bakheit AMO, Maynard VA, Curnow J, Hudson N, Kodapala S. (2003) The relation between Ashworth scale scores and the excitability of the α motor neurones in patients with post stroke spasticity. Journal of Neurology, Neurosurgery and Psychiatry 74: 646-64. Bruton A. (2002) Muscle plasticity: response to training and detraining. Physiotherapy 88(7):398-408. Fraser C et al (2002) Driving plasticity in human adult motor cortex is associated with improved motor function after brain injury. Neuron 34:831-40 Gracies JM (2006) Pathophysiology of spastic paresis: Paresis and soft tissue changes. Muscle Nerve 31: 535-551. Gracies JM. (2005) Pathophysiology of spastic paresis ll: emergence of muscle overactivity Muscle and Nerve 31:522-71. Lance J(1980) Symposium synopsis. In Feldman R, Young R, Koella W (eds) Spasticity: Disordered Motor Control. Year Book, Chicago. Jörg Wissel J, Manack A, Brainin M. (2013) Toward an epidemiology of post stroke spasticity Neurology 80: (S2) S13-S19 DOI 10.1212/WNL.0b013e3182762448. Lundstrom E, Smits A, Terent A, Borg J. (2010) Time course and determinants of spasticity during the first six months following first ever stroke Journal of Rehabilitation medicine 42: 296-301. Malhotra S, Pandyan AD, DayCR, Jones PW, Hermens H. (2009) Spasticity, an impairment that is poorly defined and poorly measured Clinical Rehabilitation 23: 651-658. Malhotra S, Cousins E, ward A, Day C, Jones P, Hoffe C, Pandyan A. (2008) An investigation into the agreement between clinical, biomechanical and neurophysiological measures of spasticity. Clinical rehabilitation 22:1105-1115. Nielsen JB, Crone C, Hultborn H. (2007) The spinal pathophysiology of spasticity – from a basic science point of view 189: 171-180. Nozaki D, Kawashima N, Aramaki Y, Akai M, Nakazawa K, Nakajima Y, Yano H. (2003) Sustained muscle contractions maintained by autonomous neuronal activity within the spinal cord. Journal of Neurophysiology 90: 2090-2097. Pandyan AD, Gregoric M, Barnes MP, Wood D et al (2005) Spasticity: clinical perceptions, neurological realities and meaningful measurement. Disability and Rehabilitation 27(1&2):2-6. Patrick T, Ada L. (2006) The Tardieu Scale differentiates contracture from spasticity whereas the Ashworth Scale is confounded by it. Clinical Rehabilitation 20: 173-182 Patten C, Lexell J, Brown HE. (2004) ‘Weakness and strength training in persons with post stroke hemiplegia: rationale, method and efficacy. J Rehabil Res Dev 41(3A):293-312. Satkunam LE. (2003) Rehabilitaion medicine: 3. Management of adult spasticity Canadian Medical Association Journal 169: 1173- 1179. Sheean G. (2002) The pathophysiology of spasticity. European Journal of Neurology 9(s1):3-9. Singer B, Dunne J, Allison G. (2001) Reflex and non-reflex elements of hypertonia in triceps surae muscles following acquired brain injury: implications for rehabilitation. Disability and Rehabilitation 23(17):749-75. Sommerfeld DK, Eek E, Svensson AK, Holmquist LW, von Arbin MH. (2004) Spasticity after stroke: Its occurrence and association with motor impairments and activity limitations 35: 134-139. © BBTA 2016 6