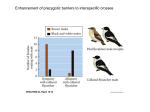
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Genomic imprinting wikipedia , lookup
Epigenetics of neurodegenerative diseases wikipedia , lookup
Biology and consumer behaviour wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Metagenomics wikipedia , lookup
Polymorphism (biology) wikipedia , lookup
Neuronal ceroid lipofuscinosis wikipedia , lookup
Genetic engineering wikipedia , lookup
Medical genetics wikipedia , lookup
Epigenetics of diabetes Type 2 wikipedia , lookup
Gene expression programming wikipedia , lookup
History of genetic engineering wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Gene expression profiling wikipedia , lookup
Frameshift mutation wikipedia , lookup
Behavioural genetics wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Genome evolution wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Human genetic variation wikipedia , lookup
Heritability of IQ wikipedia , lookup
Hardy–Weinberg principle wikipedia , lookup
Genetic drift wikipedia , lookup
Point mutation wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Genome (book) wikipedia , lookup
Designer baby wikipedia , lookup
Dominance (genetics) wikipedia , lookup
Public health genomics wikipedia , lookup
Population genetics wikipedia , lookup
Editorial Association Studies of Vascular Phenotypes How and Why? L. Almasy, J.W. MacCluer T Downloaded from http://atvb.ahajournals.org/ by guest on June 18, 2017 the disease or trait in question and the other group, controls, does not. Association exists when the allele frequencies differ between cases and controls. To avoid spurious associations, it is important that the case and control groups be matched as closely as possible for potentially confounding factors that may be correlated with the phenotype, such as ethnicity or cigarette smoking. To eliminate the need to match case and control populations, another method was developed which derives control alleles from the chromosomes carried by parents of cases.2,3 In the absence of association, there is an equal probability that either of a parent’s two alleles will be transmitted to his or her offspring. The transmission disequilibrium test, or TDT, tests whether a given allele was transmitted from a heterozygous parent to affected offspring more often than it was not transmitted. This requires that cases and both of their parents be genotyped. The genotyping of parents or other additional individuals is becoming increasingly inexpensive and helps guard against some types of spurious association. Particularly for late-onset diseases, parents of affected individuals may not be available. Extensions to the TDT have been developed which use only a single parent, siblings of affected individuals, or both parents and siblings.4,5 The most basic type of association analysis for quantitative traits involves testing whether the trait mean varies among individuals with different genotypes. This is the approach taken by Brousseau et al.1 For a rare variant, the analysis may be done by grouping subjects into two categories, those who carry the allele and those who do not. For more common polymorphisms, where all genotypes appear at appreciable frequencies, an additive model may be used. In this case, the trait mean for heterozygotes is constrained to be exactly halfway between that of the two homozygotes. This implies that each “dose” of the variant allele has the same incremental effect on phenotypic values. As with discrete traits, these tests are susceptible to hidden stratification in the sample. Although the statistical methodology is more straightforward with unrelated individuals, various methods can be used to perform this type of test in a sample of related individuals taking into account their familial relationships.6 – 8 Markers with more than two alleles complicate all of the above tests. It is common for investigators to try to reduce a multiallelic marker to a two-allele system by combining the alleles into two groups. There are many possible ways to construct these combinations, and if these permutations are not accounted for in the statistical analyses, they can greatly increase the rate of false-positive associations. Alternatively, some statistical tests have been designed specifically for use with multiallelic markers. echnological and methodological advances in the last decade have rendered genetic studies of complex traits, influenced by multiple genes and their interactions with each other and with the environment, increasingly feasible. Cardiovascular disease and its risk factors have been the subject of numerous genetic studies seeking to identify the specific DNA variants that influence these traits. One such study appears in this issue of Arteriosclerosis, Thrombosis, and Vascular Biology. Brousseau and colleagues1 investigate associations of a variant in the cholesteryl ester transfer protein (CETP) gene with lipid and lipoprotein concentrations, particle sizes, and coronary heart disease endpoints. Given the increasing frequency of genetic association studies in the pages of the Journal, we take this opportunity to explain the types of methodology used in these studies, how the results of these studies may be interpreted, and how association techniques fit into the larger arsenal of genetic epidemiological methods. See page 1148 How Do You Do an Association Study? Genetic association studies essentially look for correlations between phenotype and genotype. The phenotype may be presence or absence of disease, such as atherosclerosis, or it may be a quantitative measure such as systolic blood pressure or HDL cholesterol concentration. Slightly different analytical techniques, explained below, are used for discrete and quantitative phenotypes. The genotype is generally obtained from some type of polymorphic marker. This may be a short tandem repeat or microsatellite in which the number of copies of a 2-, 3-, or 4-base pair DNA motif varies in the population or it may be a single nucleotide polymorphism (SNP) in which a particular DNA base pair varies. A marker is said to be polymorphic and may be referred to as a polymorphism, if the frequency of the most common variant is less than 99%. Microsatellites generally have 4 to 12 variants, or alleles, whereas SNPs generally have 2. Each individual carries two alleles, one obtained from each parent. For a discrete trait, the simplest sort of association study counts the frequency of each allele at a polymorphic marker in two groups of unrelated individuals. One group, cases, has From the Department of Genetics, Southwest Foundation for Biomedical Research, San Antonio, Tex. Address correspondence to Laura Almasy, Department of Genetics, Southwest Foundation for Biomedical Research, PO Box 760549, San Antonio, TX 78245-0549. E-mail [email protected] (Arterioscler Thromb Vasc Biol. 2002;22:1055-1057.) © 2002 American Heart Association, Inc. Arterioscler Thromb Vasc Biol. is available at http://www.atvbaha.org DOI: 10.1161/01.ATV.00000024686.49995.41 1055 1056 Arterioscler Thromb Vasc Biol. July 2002 Why Might You Observe an Association? Downloaded from http://atvb.ahajournals.org/ by guest on June 18, 2017 There are three reasons why one might observe an association, or correlation, between a marker and a phenotype. It is possible that the relationship is a causal one and the genotyped marker is itself functional. This implies that the different alleles at the marker change the transcription of the DNA into RNA, affect the processing or stability of the RNA or the protein, or change the structure of the protein. A second option is that the genotyped marker is not itself functional, but is in linkage disequilibrium with other polymorphisms that are functional. Linkage disequilibrium, explained in greater detail below, is a function of both physical proximity between the marker and the functional polymorphism on the same chromosome, and their shared history in the population. Finally, it is possible that the association is due to population stratification. Population stratification refers to the case in which a correlation between a marker and a phenotype is due to each being correlated with a third, nongenetic factor. The classical example of such a factor is ethnicity. Allele frequencies differ in different ethnic groups, sometimes appreciably. If the frequency of disease also differs among these groups, an association may be observed between disease and a marker simply because the ethnic make-up of the affected and unaffected samples differs. In this case, any marker with discordant allele frequencies in the underlying populations would show an association with disease, regardless of its own functionality or its proximity to other functional polymorphisms. Given these potential sources of association, we must be cautious in the conclusions we draw from association studies. If care has been taken to avoid potential sources of stratification and to correct for multiple testing, the most likely explanation for a positive result is that one or more functional sites are in disequilibrium with the genotyped marker. If the different alleles at the genotyped marker result in changes in the amino acid structure of a protein or if previous in vitro studies have identified differences in, for example, stability, localization, binding, or transcription rate of the products of the different alleles, then we may have a stronger case for arguing that the genotyped marker is functional and directly influences the phenotype. What Is Linkage Disequilibrium? Linkage disequilibrium occurs when an allele at one genetic locus (for example, a rare mutation in a functional gene) is situated on the same chromosome with a specific allele at another locus (for example, the most common allele at a polymorphic marker locus) more often than would be expected by chance. Linkage disequilibrium between a mutation and surrounding polymorphisms is an artifact of the history of the mutation. A given mutation originates in a single individual. At that point, the new allele occurs only on that one chromosomal background, and there is complete disequilibrium between the new mutation and the surrounding markers. When this first individual reproduces, recombinations occur between the new allele and the surrounding markers. In the eggs or sperm of the original individual, the mutation now appears in connection with more than one of the alleles at the surrounding markers, though still more often on the original chromosomal background, and we have incomplete disequilibrium. The probability of recombination is proportional to the distance between the new mutation and the other marker. So markers that are closer to the new mutation are likely to be in stronger disequilibrium with it. Generations pass, more recombinations occur, and disequilibrium between the mutation and surrounding markers continually decreases. Eventually, the mutation reaches equilibrium with the surrounding markers. At equilibrium, the probability of finding a particular combination of alleles occurring together is simply the product of their individual allele frequencies. In addition to recombination, recurrence of the same mutation also decreases disequilibrium. A particular mutation may have arisen multiple times in different individuals on different chromosomal backgrounds. In this case, disequilibrium with surrounding markers may only exist in population subgroups that may not be easily identified. In association studies that seek to localize genes influencing human phenotypes, the difficulty with exploiting disequilibrium is that it is generally impossible to guess where in this process a mutation is. If a mutation influencing HDL cholesterol levels occurred only recently, then disequilibrium between it and other markers is likely to be strong. Only a small sample size will be required to detect association, and few markers will need to be genotyped because disequilibrium will extend over a broad chromosomal region. If the mutation is somewhat older, a larger sample size will be required and more markers will need to be genotyped. In some cases, when equilibrium has been reached, no disequilibrium will be present and association will not be detected unless we are lucky enough to pick the mutation itself as the marker to be genotyped for our study. Even if we are lucky enough to pick a functional site as the marker to be genotyped, we may still run into trouble if there are multiple mutations that produce similar phenotypic effects. In this case, only a few of the individuals with high cholesterol levels may carry a particular deleterious allele. Another difficulty is that the relationship between disequilibrium and distance erodes quickly. Contrary to expectation, two polymorphisms at adjacent base pairs may be in equilibrium with each other while each is in strong disequilibrium with sites tens or hundreds of base pairs away. This makes it difficult to select a subset of markers within a region that will capture the relevant genetic variation. Or, conversely, it is difficult to predict to what extent a particular set of markers represents the genetic variation within a gene or region. Tiret et al9 surveyed disequilibrium between markers in 50 candidate genes related to cardiovascular disease and found considerable variation between genes in the patterns of intragenic disequilibrium. Given this variation, the finding of negative association with a set of polymorphisms cannot exclude a particular gene, as it is possible that there are functional sites within the gene that are not in disequilibrium with the genotyped markers. Only when all polymorphic sites within a gene have been tested and rejected can we safely conclude that the gene has no effect on a phenotype. Almasy and MacCluer How Do Association Tests Fit Into the Big Picture? Downloaded from http://atvb.ahajournals.org/ by guest on June 18, 2017 With the recent progress in the human genome project and the increasing availability of sequence data and SNPs, it has been suggested that association studies will replace linkage studies as the method of choice for localizing genes influencing complex phenotypes. Linkage differs from association in that it is based on the joint transmission of a marker and a functional site from parent to offspring (ie, co-segregation), rather than on correlation. Thus, linkage studies do not require disequilibrium and are not susceptible to population stratification. However, cosegregation can only be detected by observing the passage of chromosomes between generations, and thus linkage studies require family data. On the positive side, in a linkage study, a given marker does represent a whole chromosomal region, and generalizations about a gene or a region can be made from negative results; if linkage is formally excluded in a particular chromosomal region, then we can conclude that the region does not contain genes that have a large effect on the phenotype. On the negative side, linkage exists over longer distances than disequilibrium does, and a positive result implicates an entire chromosomal region rather than a specific gene. Although the discussion of the relative merits of linkage and association methods is often framed in terms of a competition between the two, they are in fact complementary.10,11 The unpredictable nature of disequilibrium across the genome and the fact that single markers can represent entire regions in linkage studies imply that linkage methods are likely to be more efficient for initial gene localization.12 The limited extent of disequilibrium and the fact that linkage extends over large regions suggest that association methods are likely to be more useful for narrowing in on specific genes. To put it another way, linkage methods are generally good for finding new genes, and association methods are typically good for testing known ones. To screen the genome with association methods would require the genotyping of hundreds of thousands of markers, a formidable task given current technology. To screen the genome with linkage methods takes only a few hundred markers. However, linkage with markers in a candidate gene implies only that there are functional variants in that general chromosomal region, whereas association with markers in a candidate gene implies that there are functional variants very nearby. Given the family-based association methods, linkage and association analyses may be carried out in the same sample as a study progresses from initial screening to following up promising signals. Sophisticated joint tests of linkage and association can then be used to test whether polymorphisms showing Association Studies of Vascular Phenotypes 1057 association can account for a previously observed linkage signal.13 Advances in sequencing technology will soon permit investigators to carry out comprehensive surveys of the full range of polymorphisms within a candidate gene in large samples of individuals, and advances in statistical genetic methodology will provide ever more powerful techniques for extracting information from these data. Further, the current rapid development of new molecular and statistical genetic methods that can complement existing approaches will undoubtedly inspire new study designs that will increase the pace of gene discovery in the study of complex diseases. Acknowledgments The authors wish to acknowledge grants from the National Heart, Lung and Blood Institute (HL45522, HL65520, HL64244), the National Institute of Mental Health (MH59490), and the National Institute of General Medical Sciences (GM31575). References 1. Brousseau ME, O’Connor JJ Jr, Ordovas JM, Collins D, Otvos JD, Massov T, McNamara JR, Rubins HB, Robins SJ, Schaefer EJ. The CETP B2B2 genotype is associated with higher HDL cholesterol levels and lower risk for coronary heart disease end points in men with HDL deficiency. Arterioscler Thromb Vasc Biol. 2002;22:1148 –1154. 2. Falk CT, Rubinstein P. Haplotype relative risks: an easy way to construct a proper control sample for risk calculations. Ann Hum Genet. 1987;51: 227–233. 3. Spielman RS, McGinnis RE, Ewens W. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet. 1993;52:506 –516. 4. Horvath S, Laird NM. A discordant-sibship test for disequilibrium and linkage: no need for parental data. Am J Hum Genet. 1998;63:1886 –1897. 5. Sun F, Flanders WD, Yang Q, Khoury MJ. Transmission disequilibrium test (TDT) when only one parent is available: the 1-TDT. Am J Epidemiol. 1999;150:97–104. 6. Hopper JL, Matthews JD. Extensions to multivariate normal models for pedigree analysis. Ann Hum Genet. 1982;46:373–383. 7. Boerwinkle E, Chakraborty R, Sing CF. The use of measured genotype information in the analysis of quantitative phenotypes in man. Ann Hum Genet. 1986;50:181–194. 8. Zhu X, Elston RC. Transmission/disequilibrium tests for quantitative traits. Genet Epidemiol. 2001;20:57–74. 9. Tiret L, Poirier O, Nicaud V, Barbaux S, Herrmann SM, Perret C, Raoux S, Francomme C, Lebard G, Tregouet D, Cambien F. Heterogeneity of linkage disequilibrium in human genes has implications for association studies of common diseases. Hum Mol Genet. 2002;11:419 – 429. 10. Almasy L, Williams JT, Dyer TD, Blangero J. Quantitative trait locus detection using combined linkage/disequilibrium analysis. Genet Epidemiol. 1999;17(Suppl 1):S31–S36. 11. Terwilliger JD, Göring HHH. Gene mapping in the 20th and 21st centuries: statistical methods, data analysis, and experimental design. Hum Biol. 2000;72:63–132. 12. Terwilliger JD, Weiss KM. Linkage disequilibrium mapping of complex disease: fantasy or reality? Curr Opin Biotechnol. 1998;9:578 –594. 13. Soria JM, Almasy L, Souto JC, Tirado I, Borrell M, Mateo J, Slifer S, Stone W, Blangero J, Fontcuberta J. Linkage analysis demonstrates that the prothrombin G20210A mutation jointly influences plasma prothrombin levels and risk of thrombosis. Blood. 2000;95:2780 –2785. Downloaded from http://atvb.ahajournals.org/ by guest on June 18, 2017 Association Studies of Vascular Phenotypes: How and Why? L. Almasy and J.W. MacCluer Arterioscler Thromb Vasc Biol. 2002;22:1055-1057 doi: 10.1161/01.ATV.0000024686.49995.41 Arteriosclerosis, Thrombosis, and Vascular Biology is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231 Copyright © 2002 American Heart Association, Inc. All rights reserved. Print ISSN: 1079-5642. Online ISSN: 1524-4636 The online version of this article, along with updated information and services, is located on the World Wide Web at: http://atvb.ahajournals.org/content/22/7/1055 Permissions: Requests for permissions to reproduce figures, tables, or portions of articles originally published in Arteriosclerosis, Thrombosis, and Vascular Biology can be obtained via RightsLink, a service of the Copyright Clearance Center, not the Editorial Office. Once the online version of the published article for which permission is being requested is located, click Request Permissions in the middle column of the Web page under Services. Further information about this process is available in thePermissions and Rights Question and Answer document. Reprints: Information about reprints can be found online at: http://www.lww.com/reprints Subscriptions: Information about subscribing to Arteriosclerosis, Thrombosis, and Vascular Biology is online at: http://atvb.ahajournals.org//subscriptions/