Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Pharmacognosy wikipedia , lookup
NMDA receptor wikipedia , lookup
Drug interaction wikipedia , lookup
Discovery and development of TRPV1 antagonists wikipedia , lookup
Non-specific effect of vaccines wikipedia , lookup
Toxicodynamics wikipedia , lookup
Discovery and development of angiotensin receptor blockers wikipedia , lookup
5-HT2C receptor agonist wikipedia , lookup
5-HT3 antagonist wikipedia , lookup
Discovery and development of antiandrogens wikipedia , lookup
Theralizumab wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Neuropharmacology wikipedia , lookup
NK1 receptor antagonist wikipedia , lookup
Psychopharmacology wikipedia , lookup
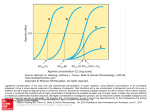
0022-3565/97/2823-1253$03.00/0 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS Copyright © 1997 by The American Society for Pharmacology and Experimental Therapeutics JPET 282:1253–1261, 1997 Vol. 282, No. 3 Printed in U.S.A. Butorphanol-Mediated Antinociception in Mice: Partial Agonist Effects and Mu Receptor Involvement1,2 H. R. GARNER,3 TIMOTHY F. BURKE,4 C. DAVID LAWHORN,5 JOANNE M. STONER and WILLIAM D. WESSINGER Department of Pharmacology and Toxicology, University of Arkansas for Medical Sciences, Little Rock, Arkansas Accepted for publication May 23, 1997 Clinically, opioids are most commonly used to provide relief of moderate-to-severe pain. The analgesics used for this purpose are predominantly mu agonists, although some effective analgesics possess significant activity at other opioid receptors. A vast number of analogs and congeners have been synthesized in an attempt to obtain compounds that retain the analgesic properties of morphine but have fewer adverse effects and lower abuse liability. Although these efforts have not resulted in the development of the “ideal opioid,” they have generated numerous unique compounds that vary widely in terms of opioid receptor-related properties such as selectivity, affinity and intrinsic efficacy. In addition to possessing substantial clinical utility, a number of these synthetic compounds have been instrumental in advancing basic Received for publication November 20, 1996. 1 A preliminary report of these findings was presented at the International Anesthesiology Research Society (IARS) meeting in Washington, DC, March 1996. 2 This study was supported by the Division of Pediatric Anesthesia, Department of Anesthesiology, University of Arkansas for Medical Sciences, Little Rock, AR, and the UAMS Graduate Student Research Fund. H.R.G. was supported by a predoctoral fellowship funded by NIDA Training Grant DA07260. 3 Present address: Johns Hopkins University School of Medicine, Behavioral Pharmacology Research Unit, 5510 Nathan Shock Dr., Suite 3000, Baltimore, MD 21224. 4 Present address: Astra Merck, Inc., 3838 N. Causeway Blvd., Lakeway III, Ste. 2400, Metairie, LA 70002. 5 Present address: 4401 Heritage Dr., Lawrence, KS 66047. effects of butorphanol in a dose-dependent insurmountable manner. Pretreatment with nor-binaltorphimine (32 mg/kg), a kappa-selective antagonist, did not reliably antagonize butorphanol, and naltrindole (20 and 32 mg/kg), a delta-selective antagonist, failed to antagonize the effects of butorphanol. Low doses of butorphanol (1.0, 1.8 or 3.2 mg/kg) caused parallel, rightward shifts in the dose-effect curve for morphine and parallel leftward shifts in the dose-effect curve for U50,488H. Taken together, the results of the present study suggest that butorphanol is a partial agonist in the mouse radiant-heat tailflick test and that activity at mu receptors accounts for the majority of its antinociceptive effects. research knowledge of opioid mechanisms. Some synthetic compounds, such as butorphanol and buprenorphine, appear to have “mixed” opioid actions; that is, they act as agonists or antagonists at multiple opioid receptors. Butorphanol is a widely used and potent analgesic with lower, although still significant, abuse potential than morphine and fentanyl (Brown, 1985; Evans et al., 1985; Smith and Davis, 1984). Over the past few years, butorphanol has gained increased clinical importance as an analgesic, a fact made clear by its recent release in a transnasal formulation (Shyu et al., 1993). Moreover, it was recently reported that butorphanol is effective in preventing the adverse side effects associated with morphine use in adults (Lawhorn et al., 1991) and in children (Lawhorn and Brown, 1994), suggesting that the antagonist actions of butorphanol can also have a clinical benefit. Butorphanol exhibits both mu and kappa agonist actions depending on the animal species and experimental conditions used (e.g., Butelman et al., 1995; Dykstra, 1990; Preston and Bigelow, 1994). In radioligand binding experiments in monkey brain, butorphanol displaces mu, kappa and delta opioids with .10-fold binding selectivity for mu vs. kappa and .30-fold mu vs. delta selectivity (Butelman et al., 1995). A similar profile of binding selectivity was observed in rodent brain (Chen et al., 1992; Horan and Ho, 1989). Moreover, butorphanol is characterized as a low efficacy or partial agonist and exhibits antagonist action at mu and kappa recep- ABBREVIATIONS: %MPE, % maximum possible effect; b-FNA, b-funaltrexamine; nor-BNI, nor-binaltorphimine; CL, confidence limit. 1253 Downloaded from jpet.aspetjournals.org at ASPET Journals on June 16, 2017 ABSTRACT In the present experiments, we characterized the agonist and antagonist effects of butorphanol in mice. In the mouse radiantheat tail-flick test, the mu agonists morphine and fentanyl and the kappa agonist U50,488H were fully effective as analgesics, whereas butorphanol was partially effective (producing 82% of maximal possible analgesic effect). Naltrexone was approximately equipotent in antagonizing the effects of morphine, fentanyl and butorphanol; in vivo apparent pA2 values for these naltrexone/agonist interactions were 7.5 (unconstrained). Naltrexone was ;10 times less potent in antagonizing the effect of U50,488H (average apparent pKB 5 6.7). The selective mu antagonist b-funaltrexamine (0.1–1.0 mg/kg) antagonized the 1254 Garner et al. in the presence of different doses of the antagonist. A less widely used analysis, the apparent pKB analysis, requires the determination of an agonist dose-effect curve in the presence of only one dose of the antagonist (Negus et al., 1993). Like the apparent pA2 analysis, the apparent pKB analysis is used to compare the potency of antagonists in blocking the effects of agonists, but it is typically used in situations in which it is not possible to redetermine an agonist dose-effect curve in the presence of multiple doses of the antagonist (e.g., limited supply of the antagonist). In the present study, these analyses are used to compare the analgesic effects of morphine, fentanyl and U50,488H with those of butorphanol in the mouse radiant-heat tail-flick test and to make inferences about the receptor population or populations that mediate these effects. To this end, the effects of the competitive antagonist naltrexone on the analgesic effects of these agonists were examined in the mouse radiant-heat tail-flick test. Another goal of the present study was to use highly specific antagonists for mu, kappa and delta receptors (i.e., b-FNA, nor-BNI or naltrindole, respectively) to further define the receptor population that mediates the antinociceptive effects of butorphanol. The development of opioid antagonists highly specific for the mu, kappa and delta receptors has played a critical role in distinguishing the individual opioid receptor types that mediate the effects of various opioids (e.g., Broadbear et al., 1994; Sofuoglu et al., 1991; Spanagel et al., 1994; Ward et al., 1982). In the mouse abdominal stretch test for antinociception, b-FNA pretreatment produced a marked rightward shift in the dose-effect curve for butorphanol, suggesting a major role for the mu receptor in its analgesic actions (Zimmerman et al., 1987). However, other investigators have classified the analgesic effects of butorphanol as being kappa mediated (e.g., Houde, 1979; Vogelsang and Hayes, 1991). Given the uncertainty surrounding the mechanisms of the analgesic action of butorphanol, we examined these effects after pretreatment with naltrexone, b-FNA, nor-BNI or naltrindole in the mouse radiant-heat tail-flick test. Finally, because it has been classified as a low efficacy agonist with antagonist properties and because the antagonist effects of butorphanol may have clinical importance, the ability of butorphanol to antagonize the analgesic effects of morphine and U50,488H was also examined. Methods Animals. The animals were experimentally naive, male ND4 Swiss-Webster mice (Harlan Sprague Dawley, Indianapolis, IN). A total of five mice were used for each point on all dose-effect curves unless otherwise indicated. At the time of use, mice weighed ;25 to 30 g. Before use, mice had unlimited access to food (PMI Feed, St. Louis, MO) and water and were housed in groups of five in a vivarium maintained on a normal phase 12-hr light/dark cycle. Apparatus and antinociception tests. An adaptation of the radiant-heat tail-flick procedure of D’Amour and Smith (1941) was used. Analgesia testing was conducted with a tail-flick apparatus (model TF-6; Emdie Instruments, Richmond, VA) that used a beam of light as the thermal nociceptive stimulus. An animal’s tail was positioned covering a photocell under the light beam. Illumination of the light started an automatic timer. The lamp was extinguished and the timer was stopped when the photocell was exposed after a mouse “flicked” its tail out of the beam. The lamp automatically extinguished after 10 sec to prevent thermal injury to the tail. The intensity of the lamp was adjustable and set so control tail-flick reaction times fell within 2 to 4 sec in control measurements. A small Downloaded from jpet.aspetjournals.org at ASPET Journals on June 16, 2017 tors (e.g. Dykstra, 1990). For example, it has been reported that in rhesus monkeys, the analgesic effects of butorphanol are mediated by mu receptors, a finding supported by its sensitivity to the antagonist effects of the competitive antagonist quadazocine and by an apparent pA2 value that has been associated with activity at mu receptors (Butelman et al., 1995). Conversely, in squirrel monkeys, butorphanol attenuates the antinociceptive effects of l-methadone (Dykstra, 1990), illustrating its antagonist actions at mu receptors. Similarly, therapeutic doses of butorphanol can cause respiratory depression (Talbert et al., 1988), yet butorphanol has also been reported to reverse the respiratory depressive effects of the selective mu agonist fentanyl (Bowdle et al., 1987). Furthermore, butorphanol increases urinary output in rats (Horan and Ho, 1989), whereas it antagonizes the increased diuretic effects of the kappa agonist bremazocine (Leander 1983a, 1983b). Interestingly, in rhesus monkeys, butorphanol is devoid of any diuretic effects, suggesting that butorphanol has no kappa agonist effects in this species (Butelman et al., 1995). Because the mixed agonist/antagonist analgesics have activity at multiple opioid receptors, the question arises as to which receptor mediates the effects of a particular opioid of this class. An important step in the pharmacological classification of the effects of an opioid compound is the evaluation of the sensitivity of these effects to antagonism by receptorselective antagonists. Several investigators have compared the potency of competitive antagonists such as naloxone, naltrexone and quadazocine in blocking various effects of opioid agonists to make inferences about mechanisms of action. For example, in a study in which the response ratedecreasing effects of various opioid agonists were examined, naltrexone is more potent as an antagonist of the effects of morphine, a mu agonist, than of ethylketocyclazocine, a kappa agonist (Harris, 1980). These results were interpreted as evidence that the rate-decreasing effects of morphine and ethylketocyclazocine are mediated by separate opioid receptor populations for which naltrexone has different affinities. Analyses of results from this type of competitive antagonism study has been broadened to include the in vivo apparent pA2 analysis (Tallarida et al., 1979). This analysis has been used to determine homogeneity of receptor populations and make inferences about agonists and antagonists with respect to the pharmacological receptor through which they produce their effects (Bertalmio and Woods, 1987; Shannon et al., 1986; Walker et al., 1994; Wessinger and McMillan, 1986). The apparent pA2 analysis provides a measure of the potency of an antagonist in blocking the effects of an agonist and is defined as the negative logarithm of the dose of the antagonist that produces a 2-fold rightward shift in the dose-effect curve of the agonist alone. The differential affinity of the competitive opioid antagonists for the mu, kappa and delta opioid receptors renders this analysis a useful tool in characterizing the opioid receptor population through which a particular effect is mediated. For example, in a study in which the antinociceptive effects of various opioid compounds in mice were examined, apparent pA2 values for naloxone in combination with mu agonists were higher than pA2 values for naloxone in combination with kappa agonists (Ward and Takemori, 1983). Determination of apparent pA2 values requires that the dose-effect curve of an agonist be redetermined several times Vol. 282 1997 Butorphanol Antinociception % MPE 5 Test reaction time 2 control reaction time 3 100 10 sec 2 Control reaction time After administration of an agonist or antagonist, if a mouse removed its tail faster than the control latency, a value of 0%MPE was assigned. Group mean 6 S.E.M. values are presented and were determined by averaging individual data from all mice tested under a given condition. A test drug was considered to be fully effective if $90%MPE was obtained. Least-squares linear regression analysis of the linear portion of the dose-effect curves was used to estimate the ED50 value, or the dose that would be expected to result in 50%MPE. The slopes of the dose-effect curves for the agonists in combination with antagonists were compared with those of the agonists alone using a parallel line assay (Tallarida and Murray, 1987). If slopes did not differ, apparent pA2 values were determined by constructing Schild plots (Arunlakshana and Schild, 1959) using drug doses instead of drug concentrations (Takemori, 1974). The apparent pA2 value represents the dose of the antagonist that would be expected to produce a 2-fold shift to the right of the dose-effect curve for the agonist alone. For Schild plot analysis, dose ratios were calculated by dividing the ED50 value of each agonist in combination with each dose of naltrexone by the ED50 for each agonist alone. The log of the dose ratios 21 (log DR 2 1) was plotted as a function of the negative log of the molar dose of the antagonist (mol/kg). A regression line was fitted to these points. Slopes of the Schild plots were considered different from unity if the 95% CL of the slope did not include 21. If the slopes of Schild regression were not significantly different from unity, the regression line was redetermined with the slope constrained to 21. The intercept of the Schild regression line on the abscissa is the apparent pA2 value. Apparent pA2 values from unconstrained and constrained Schild plots are reported here for comparison. In one instance, the slope of the regression line was determined to be statistically different from 21 (i.e., dose-effect curves were not all parallel to the initial control curves). In this case, an apparent pKB value was calculated using a modification of the equation: DR 2 1 5 B/KB (Tallarida et al., 1979), where B is the antagonist dose in mol/kg. Rearranging and taking the negative logarithm of both sides yield the following equation for determining pKB: pKB 5 2log[B/ (DR 2 1)], where DR refers to the dose ratio. The apparent pKB value is used to estimate the potency of an antagonist such as naltrexone to attenuate the effect of various agonists (Kenakin, 1987). Drugs. Morphine sulfate was obtained from Mallinkrodt (St. Louis, MO). Butorphanol tartrate was a gift from Bristol-Myers Squibb Pharmaceutical Research Institute (Princeton, NJ). Naltrexone hydrochloride and fentanyl hydrochloride were provided by the National Institute on Drug Abuse (Rockville, MD). b-FNA, nor-BNI and naltrindole, all as hydrochloride salts, were obtained from Tocris-Cookson (Langford, Bristol, UK). U50,488H (trans—(6)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)-cyclohexyl]benzeneacetamide methanesulfonate hydrate) was generously supplied by The Upjohn Co. (Kalamazoo, MI). Drugs were dissolved in 0.9% physiological saline to an injection volume of 10 ml/kg. For nor-BNI to dissolve, a drop of lactic acid was added to the solution. Doses administered are expressed as mg/kg and refer to the salt, except in the apparent pA2 analyses, in which naltrexone doses were converted to mol/kg. Results Effects of agonists alone. Morphine, fentanyl, U50, 488H and butorphanol produced dose-dependent increases in tail-flick latency (fig. 1). Maximal antinociception (i.e., .90%MPE) was observed after morphine, fentanyl and U50,488H. The dose-effect curve for butorphanol in figure 1 is the mean of two determinations [from the naltrexone antagonism studies and from the selective antagonist studies]. The ED50 (95% CL) for this mean curve was 27.6 mg/kg (19.7–38.8). Butorphanol, at the highest dose that could be administered (100 mg/kg), produced only partial analgesia (82%MPE). Doses of butorphanol of .100 mg/kg produced convulsions and were lethal within ;2 to 5 min. High doses of naltrexone (10 mg/kg) were unable to prevent this toxicity, suggesting it was a nonopioid effect. Likewise, doses of U50,488H of .180 mg/kg also produced convulsions and were lethal in all mice within ;5 to 10 min. Again, high doses of naltrexone (10 mg/kg) were unable to prevent this effect. The relative order of potency of these agonists in the mouse radiant-heat tail-flick test was fentanyl @ morphine . U50,488H $ butorphanol. Naltrexone antagonism of agonist effects. Naltrexone pretreatment (0.003–1.0 mg/kg) produced dose-dependent, parallel rightward shifts in the dose-effect curves for morphine, fentanyl and butorphanol alone (fig. 2, table 1). There was a significant difference (at P , .01; 26 df) between the slopes of the dose-effect curves for U50,488H alone and for U50,488H plus 1.0 mg/kg naltrexone; however, the slopes of the dose-effect curves for the other agonists (morphine, fentanyl or butorphanol) in combination with naltrexone were Fig. 1. Antinociceptive effects of butorphanol, morphine, fentanyl and U50,488H in the mouse radiant-heat tail-flick test. Antinociceptive effects were assessed 20 min after drug (intraperitoneally) administration and are expressed as %MPE. All points represent mean 6 S.E.M. values (n 5 5), except points on the butorphanol curve (n 5 10). The butorphanol curve is the mean of the butorphanol curves shown in figures 2 and 4. Downloaded from jpet.aspetjournals.org at ASPET Journals on June 16, 2017 number of animals (,2%) with control reaction times outside this range were excluded from the study. Mice were initially injected with saline; ;15 min later, a base-line (control) reaction time was determined. Agonist dose-effect curves were determined by the administration of fentanyl, morphine, butorphanol or U50,488H 20 min before redetermination of the tailflick reaction times. In antagonism experiments, a pretreatment dose of the antagonist (naltrexone, b-FNA, nor-BNI or naltrindole) was initially administered. After the appropriate antagonist pretreatment time (i.e., naltrexone, 20 min; naltrindole, 25 min; b-FNA or nor-BNI, 24 hr), the tail-flick reaction time was measured and then a dose of the agonist was administered. At 20 min after the agonist injection, tail-flick reaction times were measured. Injections of b-FNA, nor-BNI or naltrindole were administered subcutaneously, whereas all other agonists and antagonists were administered intraperitoneally. In other experiments, butorphanol (1.0, 1.8 or 3.2 mg/kg) was administered before morphine or U50,488H. At 15 min later, the tail-flick reaction time was measured, and then a dose of the agonist was administered. At 20 min after administration of the agonist, tail-flick reaction times were redetermined. Data analysis. The analgesic response was calculated as %MPE using the following equation: 1255 1256 Garner et al. Vol. 282 not statistically different from parallel to the dose-effect curve for the agonists alone. Doses of morphine of .560 mg/kg (e.g., 1000 mg/kg), administered after pretreatment with 1.0 mg/kg naltrexone, were lethal to all animals. Figure 3 shows the Schild plots for naltrexone as an antagonist of the antinociceptive effects of morphine, fentanyl and butorphanol. Apparent pA2 values (determined from Schild plots with the slopes unconstrained) for these interactions were 7.5. Table 2 shows the similar apparent pA2 values obtained from unconstrained and constrained Schild plot analyses. Because naltrexone did not produce consistent parallel shifts in the U50,488H dose-effect curve, apparent pA2 values were not determined. However, to obtain an estimate of the potency of naltrexone as an antagonist of the analgesic effects of U50,488H, pKB values for each dose of naltrexone (0.01, 0.1 and 1.0 mg/kg) in combination with U50,488H were calculated. Apparent pKB values for U50,488H in combination with 0.01, 0.1 or 1.0 mg/kg naltrexone were 7.4, 6.6 and 6.2, respectively, and the average apparent pKB value was 6.7. Antagonism of butorphanol effects with b-FNA, norBNI and naltrindole. Doses of 0.1, 1.0 and 10 mg/kg b-FNA administered 24 hr before butorphanol produced a dose-related antagonism of the analgesic effects of butorphanol in the mouse radiant-heat tail-flick test (fig. 4, top). Pretreatment with 0.1 mg/kg b-FNA produced a nonparallel, right- Fig. 3. Schild plots for naltrexone as an antagonist of morphine (top), fentanyl (middle) and butorphanol (bottom). Abscissae, negative logarithm of the molar doses of naltrexone (mol/kg); ordinates, logarithm of the ratio of the ED50 for the agonist in the presence of the antagonist to the ED50 of the agonist alone 21 (DR 2 1). The diagonal lines were fit to the data points by linear regression and the intercept of the fitted line is the apparent pA2 value. See text for further details. ward shift in the dose-effect curve for butorphanol alone (at P , .05, 31 df) and caused a decrease in the maximum analgesic effect produced by higher doses of butorphanol (i.e., butorphanol alone produced 82%MPE; butorphanol plus 0.1 mg/kg b-FNA produced 64%MPE). The ED50 (95% CL) values for butorphanol alone and butorphanol plus 0.1 mg/kg b-FNA were 31.9 mg/kg (20.6–57.4) and 82.3 mg/kg (60.7–136.2), respectively. Pretreatment with 1.0 mg/kg b-FNA produced a further decrease in the maximum level of analgesic effect (i.e., 37%MPE). Nearly complete antagonism of butorphanol antinociception was achieved with 10 mg/kg b-FNA pretreatment (i.e., 17%MPE). Pretreatment with 10 mg/kg nor-BNI produced a significant, ;2-fold, rightward shift in the dose-effect curve for butorphanol alone (fig. 4, center). The ED50 (95% CL) values for butorphanol alone and butorphanol plus 10 mg/kg norBNI were 31.9 mg/kg (20.6–57.4) and 74.4 mg/kg (38.6–299.7), respectively. In contrast, a higher dose of norBNI, 32 mg/kg, failed to significantly shift the dose-effect curve for butorphanol; the ED50 (95% CL) was 35.2 mg/kg TABLE 1 ED50 (95% CL) values for the analgesic effects of opioid agonists alone and in combination with naltrexone in the mouse tail-flick analgesic test Naltrexone dose Drug Alone 0.003 0.01 0.1 1.0 57.3 (34.6–95.1) 0.46 (0.28–0.73) 100.9 (74.6–161.5) 33.9 (25.9–44.2) 319.7 (241.6–462.4) 3.2 (1.8–5.4) mg/kg Morphine Fentanyl Butorphanol U50,488H 11.8 (8.7–16.7) 0.11 (0.09–0.13) 24.3 (14.2–45.8) 17.4 (12.6–23.9) 32.1 (5.2–220.4) Data are from dose-effect curves shown in figure 2. 22.6 (16.5–31.1) 0.23 (0.19–0.29) 55.0 (34.1–128.7) 22.6 (16.8–30.4) 85.2 (69.1–100.8) Downloaded from jpet.aspetjournals.org at ASPET Journals on June 16, 2017 Fig. 2. Antinociceptive effects of the agonists morphine, fentanyl, butorphanol and U50,488H alone and after administration of various doses (0.003–1.0 mg/kg) of the antagonist naltrexone (NTX). Naltrexone was administered (intraperitoneally) 20 min before administration (intraperitoneally) of the agonists. Antinociceptive effects were assessed 20 min later and are expressed as %MPE. All points represent mean 6 S.E.M. values (n 5 5, unless otherwise indicated). 1997 Butorphanol Antinociception TABLE 2 Apparent pA2 values (95% CL) and slopes for naltrexone antagonism of the antinociceptive effects of opioid agonists in the mouse tail-flick analgesic test Agonist Apparent pA2 (95% CL) Slope Apparent pA2 (95% CL) Slope of 21.0 Morphine Fentanyl Butorphanol 7.5 (6.2–8.8) 7.5 (4.2–10.9) 7.5 (5.0–9.9) 20.73 (21.5–0.04) 20.69 (22.5–1.14) 20.62 (23.0–1.8) 7.2 (6.5–7.9) 7.2 (6.4–8.1) 7.5 (6.6–8.3) Fig. 4. Antinociceptive effects of butorphanol alone and after the administration of the antagonists: b-FNA (top), nor-BNI (middle) and naltrindole (bottom). The same butorphanol dose-effect curve is depicted in each panel. The antagonists were administered (subcutaneously) 24 hr (b-FNA or nor-BNI) or 25 min (naltrindole) before administration (intraperitoneally) of butorphanol. Antinociceptive effects were assessed 20 min later and are expressed as %MPE. All points represent mean 6 S.E.M. values (n 5 5). trindole, the analgesic effects were 91.4%MPE and 78%MPE, respectively. Effects of morphine and U50,488H after low doses of butorphanol. Pretreatment with slightly effective doses of butorphanol (1.0, 1.8 or 3.2 mg/kg) produced dose-dependent, parallel, rightward shifts (antagonism) in the dose-effect curve for morphine alone (fig. 5, top). The ED50 (95% CL) values for morphine alone or morphine in combination with 1.0, 1.8 or 3.2 mg/kg butorphanol were 13.6 mg/kg (9.9–19.7), 31.6 mg/kg (23.3–44.4), 58.3 mg/kg (42.1–81.3) and 82.1 mg/kg (47.7–127.7), respectively. In contrast, pretreatment with 1.0 and 3.2 mg/kg butorphanol produced leftward, parallel shifts in the dose-effect curve for U50,488H alone, with the shift produced by the higher dose of butorphanol (3.2 mg/kg) being significantly different from control (fig. 5, bottom). The ED50 (95% CL) values for U50,488H alone and U50,488H in combination with 1.0 or 3.2 mg/kg butorphanol were 27.8 mg/kg (17.1–42.0), 23.4 mg/kg (15.7–32.2) and 17.3 mg/kg (9.2–25.6), respectively. Discussion Many of the actions of butorphanol in the mouse radiantheat tail-flick test were consistent with those of a partial mu agonist. In vivo apparent pA2 analyses showed that the interactions of naltrexone and butorphanol were similar to the interactions of naltrexone with other mu agonists. b-FNA, the selective mu antagonist, produced an insurmountable antagonism of butorphanol analgesic effects, whereas antagonists specific for kappa or delta opioids produced inconsis- Fig. 5. Effects of various doses of butorphanol administered 15 min before morphine (top) or U50,488H (bottom). Antinociceptive effects were assessed 20 min after morphine or U50,488H administration and are expressed as %MPE. All drugs were administered intraperitoneally. All points represent mean 6 S.E.M. values (n 5 5). Downloaded from jpet.aspetjournals.org at ASPET Journals on June 16, 2017 (21.7–70.9). Neither pretreatment dose of nor-BNI caused a decrease in the maximum level of analgesic effect produced by higher doses of butorphanol. The %MPE was 82% after 100 mg/kg butorphanol alone and 83% and 82% after 100 mg/kg butorphanol plus 10 or 32 mg/kg nor-BNI, respectively. Pretreatment with naltrindole (20 or 32 mg/kg) had no effect on the dose-effect curve for butorphanol alone (fig. 4, bottom). The ED50 (95% CL) values for butorphanol in combination with 20 and 32 mg/kg naltrindole were 29.8 mg/kg (17.7–44.8) and 41.5 mg/kg (28.2–61.2). Pretreatment with naltrindole also had little effect on the maximum level of antinociception produced by higher doses of butorphanol alone. The analgesic effect of 100 mg/kg butorphanol alone was 82%MPE, and in combination with 20 or 32 mg/kg nal- 1257 1258 Garner et al. nist did not have differential affinity. It is also important to note that the determination of apparent pA2 values is based on several assumptions, including that the concentrations of drugs at the receptor are directly proportional to the dose administered, agonist and antagonist interact in a competitive manner and agonist and antagonist are in equilibrium with the receptor. When the assumptions are met or approximated, the apparent pA2 analysis is fairly rigorous because it incorporates data obtained using several doses of the antagonist. Failure to meet these assumptions empirically can result in Schild plots that have slope significantly different from 21. In this case, the apparent pA2 analysis is considered inappropriate. An alternative to apparent pA2 analysis, which is useful in situations in which requirements of the Schild analysis cannot be met, is the calculation of apparent pKB values (adapted from Tallarida et al., 1979) that can also be used to estimate the potency of an antagonist such as naltrexone to antagonize the effects of various agonists. This method can be used, for example, when higher doses of the agonist produce a nonopioid-mediated toxicity, making it unfeasible to redetermine the agonist dose-effect curve in the presence of several doses of the antagonist. Comparing apparent pKB values for different agonist/antagonist interactions can implicate the involvement of a particular opioid receptors agonist effects. Because antagonists such as naltrexone are more potent in antagonizing mu-mediated effects as opposed to kappa-mediated effects, apparent pKB values for the interaction between naltrexone and a mu agonist should be higher than with a kappa agonist. Previous investigations show this to be the case. (Negus et al., 1993; Smith and Picker, 1995). Although the apparent pA2 and apparent pKB analyses both estimate antagonist potency, the apparent pA2 analysis is more stringent and provides a measure of variability. Thus, the apparent pA2 analysis should be the analysis of choice provided the previously mentioned requirements can be met. Naltrexone (0.003–1.0 mg/kg) was approximately equipotent in antagonizing the antinociceptive effects of morphine, fentanyl and butorphanol, suggesting that these agonist/antagonist interactions are mediated through the same receptor, presumably the mu receptor. Apparent pA2 values for the interactions between naltrexone and morphine, fentanyl or butorphanol were identical (i.e., 7.5) and generally agree with values obtained for naltrexone as an antagonist of mu agonists in other assays (e.g., Dykstra et al., 1988; Walker et al., 1994; Young et al.,, 1992). Furthermore, previous analysis of the naltrexone antagonism of the rate-decreasing effects of butorphanol in rats responding under a fixed ratio 30 schedule of food reinforcement yielded an apparent pA2 value of 7.2 (Pitts et al., 1996). Naltrexone (0.01, 0.1 and 1.0 mg/kg) produced dose-dependent rightward shifts in the dose-effect curve for U50,488H; however, the dose-effect curve generated by the highest dose of naltrexone in combination with U50,488H was not parallel to the dose-effect curve for U50,488H alone. Other investigators have shown that the interaction between naltrexone and U50,488H is competitive in nature and all shifts produced by increasing doses of competitive antagonists were parallel to the dose-effect curve for U50,488H alone (Dykstra, 1990; Takemori and Portoghese, 1984). In current studies, near-lethal doses of U50,488H ($100 mg/kg) were required to produce full analgesia in the mouse radiant-heat tail-flick test, thereby limit- Downloaded from jpet.aspetjournals.org at ASPET Journals on June 16, 2017 tent effects or were ineffective. The partial mu agonist effects of butorphanol were also manifested by its antagonist actions when low doses of butorphanol were combined with morphine. There was some evidence that actions of butorphanol in the present studies could also be mediated by kappa receptors. First, the selective kappa antagonist nor-BNI was effective in antagonizing the analgesic actions of butorphanol at 10 mg/kg (however, it was not effective at 32 mg/kg). Second, when low doses of butorphanol were combined with doses of U50,488H, the dose-effect curve for U50,488H shifted to the left, suggesting a coagonist effect. Butorphanol was effective as an analgesic in the mouse radiant-heat tail-flick test, but less-than-maximal analgesia was obtained. It should be noted that the inability of butorphanol to produce maximal analgesic effects may have been confounded by a ceiling effect in that doses of .100 mg/kg were lethal. In other studies with assays in which efficacy requirements are high (e.g., 55–56°C water in the tail-withdrawal test or a schedule of shock titration), butorphanol is only partially or ineffective in producing analgesia (Butelman et al., 1995; Dykstra, 1990; Morgan and Picker, 1996; O’Callaghan and Holtzman, 1975). In contrast, butorphanol produces maximum analgesia in the mouse abdominal stretch test (Zimmerman et al., 1987) and in the warm water tail-withdrawal test when lower temperatures were used (Butelman et al., 1995; Morgan and Picker, 1996). In current studies, the selective mu agonists morphine and fentanyl produced dose-related increases in antinociception and were fully effective. This is consistent with other studies using different animal models and high efficacy requiring assays (Morgan and Picker, 1996; Walker et al., 1994; Ward and Takemore, 1983). Likewise, U50,488H also produced doserelated increases in antinociception and maximal analgesia (i.e., 100%MPE was obtained). This finding is in contrast to previous investigations suggesting that kappa agonists are not as effective as mu agonists in rodent analgesia assays (Dykstra, 1985; Porreca et al., 1984; Upton et al., 1982); however, it is consistent with reports that kappa agonists are highly effective analgesics in rhesus monkeys (Dykstra et al., 1987a, 1987b). The antinociceptive effects of butorphanol were sensitive to the antagonist effects of naltrexone and permitted the use of in vivo apparent pA2 analysis for characterization of the receptor populations through which butorphanol produces its agonist effects (Dykstra et al., 1988; Shannon et al., 1986; Takemori, 1974). Classification of agonists in terms of the opioid receptor population through which they produce their effects (i.e., analgesic, discriminative-stimulus, respiratory depressive effects, etc.) is an important step in understanding tolerance and physical dependence (Feng et al., 1994; Horan and Ho, 1991), drug interactions (Dykstra, 1990; Young et al., 1992) and efficacy questions (Morgan and Picker, 1996; Picker et al., 1990). If similar pA2 values for a given antagonist against different agonists (either full or partial) are obtained, then it is considered presumptive evidence that the agonist/antagonist interactions are mediated through the same receptor population (e.g., Tallarida et al., 1979). It should be emphasized that the utility of apparent pA2 analyses in the present study depends on the differential affinity of naltrexone for mu, kappa and delta opioid receptors; this approach would not be useful to distinguish between receptors or receptor subtypes for which the antago- Vol. 282 1997 1259 of analgesia produced by butorphanol, suggesting that norBNI was not an irreversible or insurmountable antagonist of the analgesic effects of butorphanol. Previous studies using nor-BNI as an antagonist of the antinociceptive effects of mu agonists such as morphine have shown that nor-BNI does not cause a shift in the dose-effect curves for these agonists and does not decrease the level of effect obtained under control conditions (Broadbear et al., 1994; Horan et al., 1991). Finally, pretreatment with the delta-specific antagonist naltrindole (20 and 32 mg/kg) failed to antagonize the antinociceptive effects of butorphanol. In mice, naltrindole (20 mg/kg s.c.) antagonizes the antinociceptive effects of the delta-selective agonist DSLET without affecting the analgesic effects of morphine or U50,488H (Portoghese et al., 1988). Because naltrindole did not antagonize the effects of butorphanol in the present assay, it is unlikely that delta receptors play a significant role in its analgesic effects. Low doses of butorphanol in combination with morphine produced antagonist-like effects. Pretreatment with butorphanol (1.0, 1.8 or 3.2 mg/kg) caused parallel, rightward shifts in the dose-effect curve of morphine alone. This is consistent with the expectation that a low efficacy agonist would antagonize the effects of a higher efficacy agonist when given in combination. These findings are similar to those reported in a previous study in which butorphanol antagonizes the antinociceptive effects of the mu agonist l-methadone in monkeys (Dykstra, 1990). In contrast, low doses of butorphanol in combination with U50,488H produced leftward shifts in the dose-effect curve of U50,488H alone, suggesting that the antinociceptive effects of butorphanol “add to” those of U50,488H. This finding is consistent with a previous report by Butelman et al., (1995) in which butorphanol produced a nonparallel leftward shift in the antinociceptive effects of U50,488H; however, it contrasts with the Dykstra (1990) study in which butorphanol also antagonizes the analgesic effects of U50,488H, with the same potency that it antagonizes the effects of l-methadone. Butorphanol has also antagonized the effects of both mu and kappa agonists in the in vitro mouse vas deferens assay (Miller et al., 1986). The contrast in results may reflect differences in species (mouse vs. squirrel monkey) and/or experimental conditions used (thermal antinociception vs. shock titration vs. in vitro vas deferens assay). Either mu- and/or kappa-mediated antinociceptive effects of butorphanol could explain the leftward shifts of the U50,488 dose-effect curve when combined with low doses of butorphanol. For example, the data are consistent with butorphanol being an intermediate efficacy mu agonist that potentiates the antinociceptive effects of U50,488H. Alternatively, these data may reflect a summation of kappa-mediated antinociceptive effects of both drugs. In summary, most of the results presented here suggest that butorphanol acts as a partial mu agonist in producing its antinociceptive actions in the mouse radiant-heat tail-flick test. This evidence comes from pA2 analysis of naltrexone antagonism data; experiments with selective antagonists for mu, kappa and delta receptors; and the evaluation of butorphanol as an antagonist of higher-efficacy agonists. Some of the evidence also suggests that butorphanol has a kappa component to its analgesic actions in the mouse radiant-heat tail-flick test: The kappa-selective antagonist nor-BNI antagonized butorphanol analgesia, and combinations of butorpha- Downloaded from jpet.aspetjournals.org at ASPET Journals on June 16, 2017 ing the range over which the dose-effect curve could be shifted by naltrexone. This limitation of using U50,488H as an agonist in the present assay could account for this discrepancy. Because not all dose-effect curves for the naltrexone/U50,488H interaction were parallel, apparent pA2 analysis for this case was considered inappropriate. Analysis of pKB values, however, indicate that naltrexone was ;10 times less potent in antagonizing the antinociceptive effect of U50,488H than of morphine, fentanyl and butorphanol. That naltrexone produced rightward shifts in the U50,488H doseeffect curve at doses that were ;10 times the doses required to antagonize the antinociceptive effects of morphine and fentanyl suggests that U50,488H antinociception was not due to activity at mu receptors. Previous in vivo and in vitro studies show that naltrexone is much less potent in antagonizing the effects of U50,488H (Dykstra, 1990; Takemori and Portoghese, 1984). Previous studies show that b-FNA is a highly specific for mu receptors in both in vitro and in vivo assays (Takemori et al., 1981; Zimmerman et al., 1987). In the present study, b-FNA produced a dose-related antagonism of the analgesic activity of butorphanol. Pretreatment with (0.1 mg/kg) of b-FNA produced a 2.6-fold rightward shift in the dose-effect curve for butorphanol, although it was not parallel to control. This dose of b-FNA caused a “flattening” of the dose-effect curve, decreasing the maximum level of analgesia from 84%MPE to 63%MPE. Theoretically, this lack of parallelism would be expected for the interaction of full or partial agonists in combination with irreversible or insurmountable antagonists (Ruffolo, 1982). Furthermore, the lack of parallelism of the b-FNA-produced shift in the dose-effect curve of butorphanol is consistent with a previous study in which b-FNA antagonized the antinociceptive effects of various mixed agonist/antagonists, including butorphanol, in the mouse abdominal stretch test (Zimmerman et al., 1987). Results differ, however, in terms of the magnitude of the shift produced. This discrepancy can probably be explained by the much larger pretreatment dose of b-FNA (i.e., 80 mg/kg) that was used in the previous study, as well as species (rat vs. mouse) or procedural (abdominal stretch test vs. radiant-heat tail-flick test) differences. In the report by Zimmerman et al., (1987), 80 mg/kg b-FNA produces a 72.4-fold shift in the butorphanol dose-effect curve. Although the dose-effect curve generated by pretreatment with 80 mg/kg b-FNA was shifted in a nonparallel fashion, the antagonism was surmountable. Limitations imposed by the lethal effects of butorphanol in the present assay prevented the assessment of higher doses of butorphanol. The lethal effect was not prevented by a dose of 10 mg/kg naltrexone. Therefore, in the present study, it is not clear whether the antagonism of the antinociceptive effects of butorphanol by b-FNA was surmountable. Previously, nor-BNI has been reported to be a specific antagonist at kappa receptors in in vitro and in vivo studies (Broadbear et al., 1994; Portoghese et al., 1987). In the present study, pretreatment with 32 mg/kg nor-BNI failed to antagonize the analgesic effects of butorphanol; however, there was a significant rightward shift produced by pretreatment with a lower dose, 10 mg/kg nor-BNI. The reason for this inconsistent finding is presently unclear, although it suggests that some components of the analgesic effects of butorphanol are mediated through kappa receptors. Unlike b-FNA, pretreatment with nor-BNI did not cause a decrease in the maximum level Butorphanol Antinociception 1260 Garner et al. Acknowledgments The authors thank Wen-Lin Sun and Greg Davis for technical assistance and Dr. Scott Baron for helpful advice in preparation of the manuscript. References ARUNLAKSHANA, O. AND SCHILD, H. O.: Some quantitative uses of drug antagonists. Br. J. Pharmacol. 14: 48–58, 1959. BERTALMIO, A. J. AND WOODS, J. H.: Differentiation between mu and kappa receptor-mediated effects in opioid drug discrimination: Apparent pA2 analysis. J. Pharmacol. Exp. Ther. 243: 591–597, 1987. BOWDLE, T. A., GREICHEN, S. L., BJUSTROM, R. L. AND SCHOENE, R. B.: Butorphanol improves CO2 response and ventilation after fentanyl anesthesia. Anesth. Analg. 66: 517–522, 1987. BROADBEAR, J. H., NEGUS, S. S., BUTELMAN, E. R., DECOSTA, B. R. AND WOODS, J. H.: Differential effects of systemically administered nor-binaltorphimine (nor-BNI) on k-opioid agonists in the mouse writhing assay. Psychopharmacology 115: 311–319, 1994. BROWN, G. R.: Stadol dependence: Another case. JAMA 254: 910, 1985. BUTELMAN, E. R., WINGER, G., ZERNIG, G. AND WOODS, J. H.: Butorphanol: Characterization of agonist and antagonist effects in rhesus monkeys. J. Pharmacol. Exp. Ther. 272: 845–853, 1995. CHEN, J.-C., SMITH, E. R., CAHILL, M., COHEN, R. AND FISHMAN, J. B.: The opioid receptor binding of dezocine, morphine, fentanyl, butorphanol and nalbuphine. Life Sci. 52: 389–396, 1992. D’AMOUR, F. E. AND SMITH, D. L.: A method for determining loss of pain sensation. J Pharmacol. Exp. Ther. 72: 74–79, 1941. DYKSTRA, L. A.: Behavioral and pharmacological factors in opioid analgesia. In Behavioral Pharmacology: The Current Status, ed. by L. S. Seiden and R. L. Balster, pp. 111–129, Alan R. Liss, New York, 1985. DYKSTRA, L. A.: Butorphanol, levallorphan, nalbuphine and nalorphine as antagonists in the squirrel monkey. J. Pharmacol. Exp. Ther. 254: 245–252, 1990. DYKSTRA, L. A., BERTALMIO, A. J. AND WOODS, J. H.: Discriminative and analgesic effects of mu and kappa opioids: In vivo pA2 analysis. In Transduction Mechanisms of Drug Stimuli, ed. by F. C. Colpaert and R. L. Balster, pp. 107–121, Springer-Verlag, Berlin, 1988. DYKSTRA, L. A., GMEREK, D. E., WINGER, G. AND WOODS, J. H.: Kappa opioids in rhesus monkeys. I. Diuresis, sedation, analgesia and discriminative stimulus effects. J. Pharmacol. Exp. Ther. 242: 413–420, 1987a. DYKSTRA, L. A., GMEREK, D. E., WINGER, G. AND WOODS, J. H.: Kappa opioids in rhesus monkeys. II. Analysis of the antagonistic actions of quadazocine and b-funaltrexamine. J. Pharmacol. Exp. Ther. 242: 421–427, 1987b. EVANS, W. S., BOWEN, J. N., GIORDANO, F. L. AND CLARK, B.: A case of Stadol dependence. JAMA 253: 2191–2192, 1985. FENG, Y. Z., TSENG, Y. T., JAW, S. R., HOSKINS, B. AND HO, I. K.: Tolerance development to butorphanol: Comparison with morphine. Pharmacol. Biochem. Behav. 49: 649–655, 1994. HARRIS, R. A.: Interactions between narcotic agonists, partial agonists and antagonists evaluated by schedule-controlled behavior. J. Pharmacol. Exp. Ther. 213: 497–503, 1980. HORAN, P., DECOSTA, B. R., RICE, K. C. AND PORRECA, F.: Differential antagonism of U69,593- and bremazocine-induced antinociception by (2)-UPHIT: Evidence for kappa opioid receptor multiplicity in mice. J. Pharmacol. Exp. Ther. 257: 1154–1161, 1991. HORAN, P. J. AND HO, I. K.: Comparative pharmacological and biochemical studies between butorphanol and morphine. Pharmacol. Biochem. Behav. 34: 847–854, 1989. HORAN, P. J. AND HO, I. K.: The physical dependence liability of butorphanol: A comparative study with morphine. Eur. J. Pharmacol. 203: 387–391, 1991. HOUDE, R. W.: Analgesic effectiveness of the narcotic agonist-antagonists. Br. J. Clin. Pharmacol. 7: 297S–308S, 1979. KENAKIN, T. P.: Pharmacologic Analysis of Drug-Receptor Interaction, Raven Press, New York, 1987. LAWHORN, C. D. AND BROWN, R. E., JR.: Epidural morphine with butorphanol in pediatric patients. J. Clin. Anesth. 6: 91–94, 1994. LAWHORN, C. D., MCNITT, J. D., FIBUCH, E. E., JOYCE, J. T. AND LEADLEY, R. J., JR.: Epidural morphine with butorphanol for postoperative analgesia after cesarean delivery. Anesth. Analges. 72: 53–57, 1991. LEANDER, J. D.: Evidence that nalorphine, butorphanol and oxilorphan are partial agonists at a k-opioid receptor. Eur. J. Pharmacol. 86: 467–470, 1983a. LEANDER, J. D.: A kappa opioid effect: Increased urination in the rat. J. Pharmacol. Exp. Ther. 224: 89–94, 1983b. MILLER, L., SHAW, J. S. AND WHITING, E. M.: The contribution of intrinsic activity to the action of opioids in vitro. Br. J. Pharmacol. 87: 595–601, 1986. MORGAN, D. AND PICKER, M.: Contribution of individual differences to discriminative stimulus, antinociceptive, and rate decreasing effects of opioids: Importance of the drugs intrinsic efficacy at the mu receptor. Behav. Pharmacol. 7: 261–275, 1996. NEGUS, S. S., BURKE, T. F., MEDZIHRADSKY, F. AND WOODS, J. H.: Effects of opioid agonists selective for mu, kappa and delta opioid receptors on schedulecontrolled responding in rhesus monkeys: Antagonism by quadazocine. J. Pharmacol. Exp. Ther. 267: 896–903, 1993. O’CALLAGHAN, J. P. AND HOLTZMAN, S. G.: Quantification of the analgesic activity of narcotic antagonists by a modified hot-plate procedure. J. Pharmacol. Exp. Ther. 192: 497–505, 1975. PICKER, M. J., NEGUS, S. S. AND CRAFT, R. M.: Butorphanol’s efficacy at mu and kappa opioid receptors: Inferences based on schedule-controlled behavior of nontolerant and morphine-tolerant rats and on the responding of rats under a drug discrimination procedure. Pharmacol. Biochem. Behav. 36: 563–568, 1990. PITTS, R. C., WEST, J. P., MORGAN, D., DYKSTRA, L. A. AND PICKER, M. J.: Opioids and rate of positively reinforced behavior: Differential antagonism by naltrexone. Behav. Pharmacol. 7: 205–215, 1996. PORRECA, F., MOSBERG, H. I., HURST, R., HRUBY, V. J. AND BURKS, T. F.: Roles of mu, delta and kappa opioid receptors in spinal and supraspinal mediation of gastrointestinal transit effects and hot-plate analgesia in the mouse. J. Pharmacol. Exp. Ther. 230: 341–348, 1984. PORTOGHESE, P. S., LIPKOWSKI, A. W. AND TAKEMORI, A. E.: Binaltorphimine and nor-binaltorphimine: Potent and selective k-opioid receptor antagonists. Life Sci. 40: 1287–1292, 1987. PORTOGHESE, P. S., SULTANA, M. AND TAKEMORI, A. E.: Naltrindole, a highly selective and potent non-peptide d-opioid receptor antagonist. Eur. J. Pharmacol. 146: 185–186, 1988. PRESTON, K. L. AND BIGELOW, G. E.: Drug discrimination assessment of agonistantagonist opioids in humans: A three-choice saline-hydromorphonebutorphanol procedure. J. Pharmacol. Exp. Ther. 271: 48–60, 1994. RUFFOLO, R. R.: Important concepts of receptor theory. J. Auton. Pharmacol. 2: 277–295, 1982. SHANNON, H. E., CONE, E. J. AND GORODETZKY, C. W.: Morphine-like discriminative stimulus effects of buprenorphine and demethoxybuprenorphine in rats: Quantitative antagonism by naloxone. J. Pharmacol. Exp. Ther. 229: 768–774, 1986. SHYU, W. C., PITTMAN, K. A., ROBINSON, D. AND BARBHAIYA, R. H.: Multiple-dose phase I study of transnasal butorphanol. Clin. Pharmacol. Ther. 54: 34–41, 1993. SMITH, M. A. AND PICKER, M. J.: Examination of the kappa agonist and antagonist properties of opioids in the rat drug discrimination procedure: Influence of training dose and intrinsic efficacy. Behav. Pharmacol. 6: 703–717, 1995. SMITH, S. G. AND DAVIS, W. M.: Nonmedical use of butorphanol and diphenhydramine. JAMA 252: 1010, 1984. SOFUOGLU, M., PORTOGHESE, P. S. AND TAKEMORI, A. E.: Differential antagonism of delta opioid agonists by naltrindole and its benzofuran analog (NTB) in mice: Evidence for delta opioid receptor subtypes. J. Pharmacol. Exp. Ther. 257: 676–680, 1991. SPANAGEL, R., ALMEIDA, O. F. X. AND SHIPPENBERG, T. S.: Evidence that norbinaltorphimine can function as an antagonist at multiple opioid receptor subtypes. Eur. J. Pharmacol. 264: 157–162, 1994. TAKEMORI, A. E.: Determination of pharmacological constants: Use of narcotic antagonists to characterize analgesic receptors. In Narcotic Antagonists, ed. by M. C. Braude, L. S. Harris, E. L. May, J. P. Smith and J. E. Villarreal, Advances in Biochemical Psychopharmacology, Vol. 8, pp. 335–344, Raven Press, New York, 1974. TAKEMORI, A. E., LARSON, D. L. AND PORTOGHESE, P. S.: The irreversible narcotic antagonistic and reversible agonistic properties of the fumaramate methyl ester derivative of naltrexone. Eur. J. Pharmacol. 70: 445–451, 1981. TAKEMORI, A. E. AND PORTOGHESE, P. S.: Comparative antagonism by naltrexone and naloxone of m, k, and d agonists. Eur. J. Pharmacol. 104: 101–104, 1984. Downloaded from jpet.aspetjournals.org at ASPET Journals on June 16, 2017 nol with U50,488H produces greater antinociception than U50,488H alone, suggesting a coagonist action. That butorphanol may have kappa activity in this assay was not surprising given that numerous studies (some of which are reviewed herein) have reported a kappa component to the actions of butorphanol. What was surprising was that the data supporting a kappa-antinociceptive effect is somewhat equivocal. In the selective antagonist study, the antagonism of butorphanol analgesia by nor-BNI was not dose dependent. Furthermore, one could interpret the coagonist effects of butorphanol in combination with U50,488H as the summed actions of a mu (butorphanol) and kappa (U50,488H) agonist. However, a common theme throughout the butorphanol literature is that results often seem to depend on species and the particular effect being measured. Taken together, the results of the present study suggest that butorphanol acts as a partial agonist in the mouse radiant-heat tail-flick test and that activity at mu receptors accounts for the majority of its antinociceptive effects. Vol. 282 1997 TALBERT, R. L., PETERS, J. I., SORRELS, S. C. AND SIMMONS, R. S.: Respiratory effects of high-dose butorphanol. Acute Care 12: (suppl. 1) 47–56, 1988. TALLARIDA, R. J., COWAN, A. AND ADLER, M. W.: pA2 and receptor differentiation: A statistical analysis of competitive antagonism. Life Sci. 25: 637–654, 1979. TALLARIDA, R. J. AND MURRAY, R. B.: Manual of Pharmacologic Calculations with Computer Programs, 2nd ed., Springer-Verlag, New York, 1987. UPTON, N., SEWELL, R. D. E. AND SPENCER, P. S. J.: Differentiation of potent mand k-opiate agonists using heat and pressure antinociceptive profiles and combined potency analysis. Eur. J. Pharmacol. 78: 421–429, 1982. VOGELSANG, J. AND HAYES, S. R.: Butorphanol tartrate (Stadol): A review. J. Post Anesth. Nursing 6: 129–135, 1991. WALKER, E. A., MAKHAY, M. M., HOUSE, J. D. AND YOUNG, A. M.: In vivo apparent pA2 analysis for naltrexone antagonism of discriminative stimulus and analgesic effects of opiate agonists in rats. J. Pharmacol. Exp. Ther. 271: 959–968, 1994. WARD, S. J., PORTOGHESE, P. S. AND TAKEMORI, A. E.: Pharmacological characterization in vivo of the novel opiate, b-funaltrexamine. J. Pharmacol. Exp. Ther. 220: 494–498, 1982. WARD, S. J. AND TAKEMORI, A. E.: Relative involvement of mu, kappa and delta Butorphanol Antinociception 1261 receptor mechanisms in opiate-mediated antinociception in mice. J. Pharmacol. Exp. Ther. 224: 525–530, 1983. WESSINGER, W. D. AND MCMILLAN, D. E.: Quantitative analysis of naloxone antagonism of the discriminative stimulus properties of morphine in the pigeon. Pharmacol. Biochem. Behav. 25: 209–214, 1986. YOUNG, A. M., MASAKI, M. A. AND GEULA, C.: Discriminative stimulus effects of morphine: Effects of training dose on agonist and antagonist effects of mu opioids. J. Pharmacol. Exp. Ther. 261: 246–257, 1992. ZIMMERMAN, D. M., LEANDER, J. D., REEL, J. K. AND HYNES, M. D.: Use of b-funaltrexamine to determine mu opioid receptor involvement in the analgesic activity of various opioid ligands. J. Pharmacol. Exp. Ther. 241: 374– 378, 1987. Send reprint requests to: Dr. William D. Wessinger, University of Arkansas for Medical Sciences, Department of Pharmacology and Toxicology, Slot 611, 4301 W. Markham Street, Little Rock, AR 72205. E-mail: wdwessinger@ life.uams.edu Downloaded from jpet.aspetjournals.org at ASPET Journals on June 16, 2017