Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Light-dependent reactions wikipedia , lookup
Nucleic acid analogue wikipedia , lookup
Photosynthesis wikipedia , lookup
Point mutation wikipedia , lookup
Proteolysis wikipedia , lookup
Peptide synthesis wikipedia , lookup
Mitochondrion wikipedia , lookup
Nicotinamide adenine dinucleotide wikipedia , lookup
Genetic code wikipedia , lookup
Metalloprotein wikipedia , lookup
Specialized pro-resolving mediators wikipedia , lookup
Microbial metabolism wikipedia , lookup
Butyric acid wikipedia , lookup
Adenosine triphosphate wikipedia , lookup
Evolution of metal ions in biological systems wikipedia , lookup
Oxidative phosphorylation wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Basal metabolic rate wikipedia , lookup
Biosynthesis wikipedia , lookup
Glyceroneogenesis wikipedia , lookup
Fatty acid synthesis wikipedia , lookup
Fatty acid metabolism wikipedia , lookup
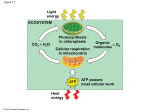
Perspectives in Nutrition, 8th Edition Chapter 9 Outline: Energy Metabolism After studying this chapter, you will be able to: 1. Explain the differences among metabolism, catabolism, and anabolism. 2. Describe aerobic and anaerobic metabolism of glucose. 3. Illustrate how energy is extracted from glucose, fatty acids, amino acids, and alcohol using metabolic pathways, such as glycolysis, beta-oxidation, the citric acid cycle, and the electron transport system. 4. Describe the role that acetyl-CoA plays in cell metabolism. 5. Identify the conditions that lead to ketogenesis and its importance in survival during fasting. 6. Describe the process of gluconeogenesis. 7. Discuss how the body metabolizes alcohol. 8. Compare the fate of energy from macronutrients during the fed and fasted states. 9. Describe common inborn errors of metabolism. 9.1 Metabolism: Chemical Reactions in the Body A. General 1. Metabolism: entire network of chemical processes involved in maintaining life a. Release of energy from macronutrients b. Biosynthesis c. Prepare wastes for excretion 2. Metabolic pathway: group of biochemical reactions that occur in sequence a. Anabolic: use small, simple compounds to build large, complex compounds; growth b. Catabolic: break down compounds into smaller units; weight loss or wasting c. In general, balance exists between anabolism and catabolism 3. Intermediates: compounds formed within a metabolic pathway B. Energy for the Cell 1. Uses a. Anabolism b. Muscle contraction c. Nerve conduction d. Ion pumps 2. Sources: catabolic reactions that break chemical bonds in carbohydrate, fat, protein, and alcohol (originally produced during photosynthesis) to form ATP, heat, carbon dioxide, and water 3. Adenosine Triphosphate (ATP) a. Main form of energy used by body b. Structure: adenosine bound to 3 phosphate groups c. The bonds between adenosine and phosphate groups contain energy d. C. 9.2 To release energy from ATP, hydrolysis of one phosphate yields adenosine diphosphate (ADP) plus a free phosphate group, then adenosine monophosphate (AMP) + Pi e. ATP is constantly recycled within cells i. Body contains 0.22 lb (100 g) of ATP ii. Sedentary adult uses 88 lb (40 kg) of ATP/d iii. During 1 hour of strenuous exercise, 66 lb (30 kg) of ATP are used Oxidation-Reduction Reactions: Key Processes in Energy Metabolism 1. Oxidation-reduction reactions transfer electrons and hydrogen ions from energyyielding compounds to oxygen, forming water and energy for ATP synthesis from ADP + Pi 2. Oxidation and reduction occur together a. Inorganic substance is oxidized: loss of 1 or more electrons (LEO) b. Inorganic substance is reduced: gain of 1 or more electrons (GER) c. Organic substance is oxidized: gain of oxygen or loss of hydrogen; hydrogen atoms are eventually added to oxygen to form water d. Organic substance is reduced: loss of oxygen or gain of hydrogen 3. Oxidation-reduction reactions are controlled by enzymes (e.g., dehydrogenases) 4. Coenzymes involved in dehydrogenase reactions a. Nicotinamide adenine dinucleotide (contains niacin) i. Oxidized form: NAD+ ii. Reduced form: NADH b. Flavin adenine dinucleotide (contains riboflavin) i. Oxidized form: FAD ii. Reduced form: FADH2 ATP Production from Carbohydrates A. General 1. Energy stored in foods is trapped in ATP or released as heat (maintains body temperature) 2. ATP is generated through cellular respiration: oxidation of food molecules to obtain energy; oxygen is final electron acceptor a. Aerobic: oxygen is readily available; cellular respiration is more efficient (net gain of 30 - 32 ATP from glucose) b. Anaerobic: oxygen is not present; cellular respiration is less efficient (net gain of 2 ATP from glucose) B. Glycolysis (see Figure 9-7) 1. Break down carbohydrates to generate energy: 1 glucose (6 carbons) is oxidized to form 2 pyruvate (3 carbons), NADH + H+, and net of 2 ATP 2. Provide building blocks for synthesis of other compounds 3. Occurs in cytosol C. Synthesis of Acetyl CoA (see Figure 9-7) Pyruvate is oxidized and joined with CoA to form acetyl-CoA, NADH + H+, and CO2 2. Catalyzed by pyruvate dehydrogenase 3. Irreversible transition reaction 4. Occurs in mitochondria 5. Requires four B-vitamins a. Thiamin b. Riboflavin: FADH or FADH2 c. Niacin: NAD+ or NADH d. Panthothenic acid: precursor to CoA Citric Acid Cycle (see Figure 9-8) 1. Alternate names for citric acid cycle a. Tricarboxylic acid (TCA) cycle b. Krebs cycle 2. Acetyl-CoA leads to production of NADH + H+, FADH2, ATP, and CO2 3. Occurs in mitochondria 4. Does not require oxygen 5. Two cycles per glucose molecule 6. Each cycle yields a. 2 CO2 b. 1 GTP (potential for ATP) c. 3 NADH + H+ d. 1 FADH2 Electron Transport Chain (see Figure 9-10) 1. NADH + H+ are oxidized to NAD+, and FADH2 is oxidized to FAD to produce ATP and water 2. Occurs in mitochondria 3. Produces 90% of ATP from glucose 4. As electrons are passed from one carrier to the next, small amounts of energy are released (oxidative phosphorylation) 5. Requires minerals a. Copper: component of enzyme b. Iron: component of cytochromes (electron-transfer compound) The Importance of Oxygen 1. NADH + H+ and FADH2 from citric acid cycle are regenerated to NAD+ and FAD by transfer of their electrons and hydrogen to oxygen in the electron transport chain 2. Without oxygen, cells would not be able to generate sufficient energy to sustain life Anaerobic Metabolism 1. For cells that are not capable of aerobic respiration (lack mitochondria; e.g., RBCs) or need to produce energy in the absence of oxygen (e.g., during intense 1. D. E. F. G. H. 9.3 exercise), pyruvate may be converted to lactate, providing only 5% of potential ATP (2 ATP) 2. Transfer of hydrogen from NADH + H+ to pyruvate produces lactate + NAD+ 3. NAD+ continues function of glycolysis 4. Lactate is released into bloodstream, picked up by liver, and used to synthesize pyruvate, glucose, or some other intermediate in aerobic respiration 5. Buildup of lactate in exercising muscle makes it difficult for muscles to contract Overall energy yield of aerobic respiration 1. Each NADH yields 2.5 ATP 2. Each FADH2 yields 1.5 ATP 3. Total ATP from each glucose molecule = 32 ATP a. Glycolysis 2 ATP b. Citric acid cycle (GTP) 2 ATP c. Citric acid cycle (ATP) 28 ATP ATP Production from Fats A. General 1. Lipolysis: breakdown of triglycerides into free fatty acids and glycerol 2. Fatty acid oxidation: breakdown of fatty acids for energy production; net donation of electrons from fatty acids to oxygen; takes place in mitochondria 3. Sources of fatty acids for oxidation a. Dietary fat (e.g., following high-fat meal) b. Fat stored in adipose tissue (e.g., in fasting state) 4. Release of fat from adipose cells relies on hormone-sensitive lipase 5. Hormone-sensitive lipase activity is affected by: a. Glucagon (increases HSL activity) b. Growth hormone (increases HSL activity) c. Epinephrine (increases HSL activity) d. Insulin (decreases HSL activity) 6. Fatty acids are shuttled from cytosol into mitochondria by carnitine B. ATP Production from Fatty Acids 1. Most fatty acids in nature have even number of carbons (2 to 26) 2. Beta-oxidation: 2-carbon fragments from a fatty acid are cleaved and converted to acetyl-CoA (see Figure 9-13) 3. Acetyl-CoA enters citric acid cycle to produce CO2 and ATP a. Glucose yields 30 - 32 ATP (5 ATPs/carbon) b. 16-carbon fatty acid yields 104 ATP (7 ATPs/carbon, due to more carbon-hydrogen bonds) 4. A fatty acid with an odd number of carbons forms a 3-carbon compound (propionyl-CoA), which bypasses the acetyl-CoA step when it enters the citric acid cycle to yield NADH + H+ and FADH2 or can be used to synthesize other products (e.g., glucose) C. Carbohydrate Aids Fat Metabolism 1. D. E. F. 9.4 Some intermediates of citric acid cycle enter other biosynthetic pathways, which depletes oxaloacetate supply 2. Formation of pyruvate from carbohydrate metabolism replenishes oxaloacetate Ketogenesis 1. Ketone bodies: products of incomplete fatty acid oxidation, mostly resulting from hormonal imbalances (e.g., inadequate insulin production) a. Acetoacetic acid b. Beta-hydroxybutyric acid c. Acetone 2. Ketosis: excess of ketone bodies (see Figure 9-15) a. Decreases pH of body fluids b. Characterized by fruity breath odor (from excretion of acetone via lungs) 3. Most ketone bodies are converted back into acetyl-CoA and metabolized via citric acid cycle Ketosis in Diabetes 1. Lack of insulin inhibits normal carbohydrate and fat metabolism 2. Cells cannot utilize glucose, so fatty acids are rapidly metabolized to ketone bodies 3. Excessive ketones are excreted in urine along with sodium and potassium, leading to ion imbalances 4. Diabetic ketoacidosis (typically only seen with uncontrolled type 1 diabetes) a. pH of blood decreases b. May lead to coma or death c. Treatment: insulin, electrolytes, fluids Ketosis in Semistarvation or Fasting 1. Low insulin production during semistarvation or fasting causes fatty acids to flood the bloodstream and form ketone bodies in the liver 2. Ketone bodies may be used for fuel by heart, muscles, kidneys, and brain (after several days) 3. As cells adapt to using ketones for fuel, less glucose is produced from amino acids (sparing protein) 4. Death occurs after 50 - 70 days of total fasting, when about half of body protein is depleted Protein Metabolism A. General 1. Metabolism of protein takes place primarily in the liver 2. Muscle metabolism of protein is limited to branched-chain amino acids (leucine, isoleucine, and valine) 3. Deamination: removal of amino group from amino acid; often requires vitamin B-6 4. Produces carbon skeletons that serve as intermediates in citric acid cycle or yield acetyl-CoA or pyruvate B. C. D. Gluconeogenesis: Producing Glucose from Glucogenic Amino Acids and Other Compounds 1. Gluconeogenesis: production of glucose from certain amino acids in liver and some kidney cells 2. In the mitochondria, carbon skeleton of a glucogenic amino acid is converted to oxaloacetate 3. Oxaloacetate moves to cytosol, loses 1 CO2, becomes phosphoenolpyruvate 4. Phosphoenolpyruvate reverses the path through glycolysis to glucose 5. Gluconeogenesis requires ATP, biotin, riboflavin, niacin, and vitamin B-6 Gluconeogenesis from Typical Fatty Acids is Not Possible 1. Fatty acids with even number of carbons are broken down to acetyl-CoA 2. The reaction that converts pyruvate to acetyl-CoA is irreversible 3. Acetyl-CoA may only form ketones and/or combine with oxaloacetate in the citric acid cycle 4. During citric acid cycle, acetyl-CoA is added to oxaloacetate, but 2 carbons are lost as CO2; thus, no carbons from acetyl-CoA remain to form glucose 5. Glycerol portion of triglyceride may be converted to glyceraldehyde 3-phosphate to yield glucose; yield is insignificant Disposal of Excess Amino Groups from Amino Acid Metabolism 1. Amino groups (-NH2) are converted to ammonia (NH3),which is toxic to cells 2. In the liver, ammonia is converted to urea by the urea cycle a. Urea is excreted via urine 3. Nitrogen in blood is a sign of organ disease a. High [NH3] = liver disease b. High [urea] = kidney disease 9.5 Alcohol Metabolism (see Figure 9-18) A. Alcohol dehydrogenase (ADH) pathway is primary pathway for alcohol metabolism 1. Alcohol acetaldehyde by alcohol dehydrogenase; yields NADH + H+ 2. Acetealdehyde acetyl-CoA by aldehyde dehydrogenase; yields NADH + H+ 3. Occurs mainly in liver, although 10 - 30% may occur in stomach lining cells 4. Fate of acetyl-CoA a. Some may enter citric acid cycle, but ADH pathway limits supply of NAD+; ADH pathway takes priority over citric acid cycle in cells b. Most is directed to fatty acid and triglyceride synthesis i. Results in steatosis: accumulation of fat in the liver ii. Characterized by high blood triglycerides B. Microsomal ethanol oxidizing system (MEOS) metabolizes alcohol intake in excess of capacity of ADH pathway C. Catalase pathway is minor D. Exceeding capacity of alcohol metabolizing systems leads to alcohol poisoning 9.6 Regulation of Energy Metabolism A. B. C. D. Overview of metabolic pathways 1. Glycolysis (glucose pyruvate) in cytosol 2. Transition reaction (pyruvate acetyl-CoA) in mitochondria 3. Fatty acid oxidation (fatty acid acetyl CoA) in mitochondria 4. Glucogenic amino acid oxidation (amino acids pyruvate) in cytosol 5. Non-glucogenic amino acid oxidation (amino acids acetyl-CoA) in mitochondria 6. Alcohol oxidation (ethanol acetaldehyde) in cytosol 7. Alcohol oxidation (acetaldehyde acetyl-CoA) in mitochondria 8. Citric acid cycle (acetyl-CoA CO2) in mitochondria 9. Gluconeogenesis in mitochondria and cytosol The Liver (see Figure 9-20) 1. Conversions between various simple sugars 2. Fat synthesis 3. Production of ketone bodies 4. Amino acid metabolism 5. Urea production 6. Alcohol metabolism 7. Nutrient storage ATP Concentrations 1. High [ATP] decreases energy-yielding reactions and increases anabolic reactions 2. High [ADP] increases energy-yielding reactions and decreases anabolic reactions Enzymes, Hormones, Vitamins, and Minerals 1. Enzymes a. Presence (i.e., enzyme synthesis) b. Rate of activity 2. Hormones a. Regulate metabolic processes b. Low [insulin] promotes gluconeogenesis, protein breakdown, and lipolysis c. High [insulin] promotes synthesis of glycogen, fat, and protein 3. Vitamins and minerals (see Figure 9-21) a. Thiamin b. Riboflavin c. Niacin d. Pantothenic acid e. Biotin f. Vitamin B-6 g. Folate h. Vitamin B-12 i. Iron j. Copper 9.7 Fasting and Feasting A. Fasting 1. Postprandial fasting (0 - 6 hours after eating) a. Use of liver stores of carbohydrate (glycogen) b. Use of adipose stores of fatty acids 2. Short-term fasting (3 - 5 days) a. Glycogen stores are depleted b. Fatty acid oxidation continues c. Gluconeogenesis (amino acids from lean tissue and glycerol from triglycerides glucose) to provide fuel for CNS and RBCs i. Early gluconeogenesis leads to rapid breakdown of lean tissue (supplies 90% of glucose) that would lead to death within 2- 3 weeks ii. Sodium and potassium are also depleted via urine when ketone bodies are excreted iii. Blood urea levels increase 3. Long-term fasting (5+ days) a. Metabolic rate slows, reduces energy requirements b. CNS begins to use ketone bodies c. When lean body mass declines by 50% (7 - 10 weeks), death occurs B. Feasting 1. Accumulation of body fat 2. Insulin production encourages use of glucose for energy, synthesis of glycogen, and storage of protein and fat 3. Fat consumed in excess of need is stored in adipose cells (requires little energy) 4. Protein consumed in excess of need does not promote muscle development a. Some remains in blood’s amino acid pool (minor) b. Some used to synthesize fatty acids (minor; costly process requires ATP, biotin, niacin, and pantothenic acid) c. Some metabolized for energy 5. Carbohydrate consumed in excess of need a. Maximize glycogen stores b. Used as fuel, lessens need for fatty acid oxidation c. Storage as fat (not an active pathway in humans; uses ATP, biotin, niacin, and pantothenic acid) 6. Synthesis of fat from carbohydrate or protein (lipogenesis) takes place in cytosol of liver cells a. Acetyl-CoA from glucose or amino acids is linked into palmitic acid (16carbon fatty acid) b. Insulin increases activity of fatty acid synthase, which regulates lipogenesis c. Palmitic acid can be lengthened to 18- or 20-carbon chain d. Used to synthesize triglycerides 9.8 Medical Perspective: Inborn Errors of Metabolism A. General 1. Lack of enzyme to perform a specific metabolic function 2. Products of disrupted metabolism may be toxic to the body 3. Inherit defective gene from one or both parents (may not have the disease, but are carriers) 4. Symptoms of disorder appear shortly after birth (e.g., loss of appetite, vomiting, dehydration, weakness, developmental delays) 5. Specific - involve 1 or a few enzymes 6. No cure, only treatment B. Newborn Screening 1. Public health program that provides early identification and follow-up treatment of infants with genetic and metabolic disorders 2. Components of screening are state-specific C. Phenylketonuria (PKU) 1. 1/13,500 -19,000 births 2. Most common among people of Irish descent 3. Carriers are detected by simple blood test 4. Newborn screening for PKU is required in all states 5. Inefficient function of phenylalanine hydroxylase in liver 6. Treatment a. Phenylalanine-restricted diet (phenylalanine cannot be omitted because it is an essential amino acid) b. Continual monitoring of infants through blood testing c. Specialized infant formulas are needed to meet high protein needs without high phenylalanine d. Foods with low phenylalanine content i. Fruits ii. Vegetables e. Foods with moderate phenylalanine content i. Breads ii. Cereals f. Foods with high phenylalanine content i. Dairy products ii. Eggs iii. Meats iv. Nuts v. Cheeses vi. Aspartame (artificial sweetener made with phenylalanine) g. Older children and adults must still rely on low-phenylalanine formula 7. Low-phenylalanine diet must be continued throughout life or decreased intelligence and behavioral problems occur 8. D. E. During pregnancy, exposure to toxic levels of phenylalanine may lead to miscarriage or birth defects Galactosemia 1. 1/47,000 births 2. Most common among people of Italian and Irish descent 3. 2 specific enzyme defects lead to reduced metabolism of galactose to glucose; buildup of galactose can lead to serious bacterial infections, mental retardation, and cataracts 4. Infants with galactosemia develop vomiting after a few days of infant formula or breast milk 5. Treatment a. Soy formula b. Avoidance of certain foods i. Lactose-containing products ii. Organ meats iii. Some fruits and vegetables c. Strict label reading Glycogen Storage Disease 1. 1/60,000 births 2. Inability to convert glycogen to glucose due to one of a number of enzyme defects 3. Symptoms a. Poor growth b. Low blood glucose c. Liver enlargement 4. Treatment a. Frequent meals to regulate blood glucose b. Raw cornstarch (slowly digested, maintains blood glucose) c. Careful monitoring of blood glucose