Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Paracrine signalling wikipedia , lookup
Ultrasensitivity wikipedia , lookup
Signal transduction wikipedia , lookup
Gene expression wikipedia , lookup
Photosynthetic reaction centre wikipedia , lookup
Point mutation wikipedia , lookup
Mitogen-activated protein kinase wikipedia , lookup
NADH:ubiquinone oxidoreductase (H+-translocating) wikipedia , lookup
Expression vector wikipedia , lookup
Magnesium transporter wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
Evolution of metal ions in biological systems wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Structural alignment wikipedia , lookup
Western blot wikipedia , lookup
Deoxyribozyme wikipedia , lookup
Protein purification wikipedia , lookup
Interactome wikipedia , lookup
Homology modeling wikipedia , lookup
Proteolysis wikipedia , lookup
Catalytic triad wikipedia , lookup
Two-hybrid screening wikipedia , lookup
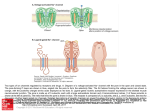
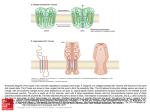
Chapter 86 The Structure and Topology of Protein Serine/Threonine Phosphatases David Barford Section of Structural Biology, Institute of Cancer Research, Chester Beatty Laboratories, London, England, UK Introduction Structural studies of the two families of protein phosphatases responsible for dephosphorylating serine and threonine residues have revealed that, although these families are unrelated in sequence, the architecture of their catalytic domains is remarkably similar and distinct from the protein tyrosine phosphatases. The diversity of structure within the PPP and PPM families is generated by regulatory subunits and domains that function to modulate protein specificity and to localize the phosphatase to particular subcellular locations [1]. Protein serine/threonine phosphatases of the PPP family The protein Ser/Thr phosphatases PP1, PP2A, and PP2B of the PPP family, together with PP2C of the PPM family, account for the majority of the protein serine/threonine phosphatase activity in vivo. While PP1, PP2A, and PP2B share a common catalytic domain of 280 residues, these enzymes are divergent within their non-catalytic N and C termini and are distinguished by their associated regulatory subunits to form a diverse variety of holoenzymes. Major members of the PPP family are encoded by numerous isoforms that share a high degree of sequence similarity, especially within their catalytic domains. Greater sequence diversity occurs within the extreme N and C termini of the proteins. Although these isoforms have similar substrate specificities and interact with the same regulatory subunits in vitro, the phenotype of a functional loss is isoform specific, indicating they perform distinct functions in vivo. Handbook of Cell Signaling, Three-Volume Set 2 ed. Copyright © 2010 Elsevier Inc. All rights reserved. Overall Structure and Catalytic Mechanism Structural analyses of members of the PPP family have begun to reveal the molecular basis for catalysis, inhibition by toxins, and aspects of their regulatory mechanisms. Crystal structures are available for (1) various isoforms of PP1 in complex with natural toxins [2, 3], the phosphate mimic tungstate [4], and a peptide of the RVxF targeting motif [5]; (2) PP2B in the auto-inhibited state [6] and as a complex with FK506/FKBP [6, 7]; (3) the PR65/A-subunit of PP2A [8]; and (4) a regulatory TPR domain of PP5 [9]. The catalytic domains of the PP1 catalytic subunit (PP1c) and PP2B share a common architecture consisting of a central -sandwich of two mixed -sheets surrounded on one side by seven -helices and on the other by a subdomain comprised of three -helices and a three-stranded -sheet (Figure 86.1a). The interface of the three -sheets at the top of the -sandwich creates a shallow catalytic site channel. Conserved amino acid residues present on loops emanating from the -strands of this central -sandwich are responsible for coordinating a pair of metal ions to form a binuclear metal centre (Figure 86.2a). Crystallographic data on PP1c and PP2B provided the first compelling insight regarding the role of metal ions in the catalytic reaction of the PPP family. The identity of the two metal ions is slightly controversial. Proton-induced X-ray emission spectroscopy performed on PP1c crystals produced from the protein expressed in Escherichia coli indicated that the metal ions were Fe2 (or Fe3) and Mn2 [4], whereas atomic absorption spectroscopy of bovine brain PP2B indicated a stoichiometric ratio of Zn2 and iron [10]. There have also been conflicting reports concerning the iron oxidation states, with both Fe2 and Fe3 being observed. Native 677 678 Figure 86.1 Ribbon diagrams showing an overview of the structures of protein Ser/Thr phosphatase catalytic domains. (a) The catalytic subunit of human protein phosphatase 1 in complex with the RVxF motif PP1 binding peptide. The catalytic site is indicated by the two metal ions and bound sulfate ion. (b) Human PP2C the catalytic site is indicated by the two metal ions and bound phosphate ion. Figure 86.1a reproduced from [5] (Egloff et al., 1997), with permission; Figure 86.2 is reproduced from [19] (Das et al., 1996), with permission. PP2B is most likely to contain Fe2, explaining the timedependent inactivation of PP2B that results from the oxidation of the Fe–Zn center [11]. Oxidation of the binuclear metal center and phosphatase inactivation may represent a mechanism for PP2B regulation by redox potential during oxidative stress or as a result of reactive oxygen species generation following receptor tyrosine kinase activation, in a process reminiscent of the inactivation of protein tyrosine phosphatases by oxidation of the catalytic site Cys residue by hydrogen peroxide and perhaps analogous to a possible PP2C regulatory mechanism, discussed later [10]. The structure of PP1c with tungstate and PP2B with phosphate indicated that two oxygen atoms of the oxyanion-substrate coordinate the metal ions (Figure 86.2a) [4, 7]. Two water molecules, one of which is a metalbridging water molecule, contribute to the octahedral hexa-coordination of the metal ions. The metal coordinating residues (aspartates, histidines, and asparagines) are PART | II Transmission: Effectors and Cytosolic Events Figure 86.2 Comparison of the catalytic sites of the protein Ser/Thr phosphatases, showing the common binuclear metal center, coordinating the phosphate of the phosphorylated protein substrate: (a) human PP1 (for clarity, Arg 96 is not shown) and (b) human PP2C. invariant among all PPP family members. These residues, together with Arg and His residues that interact with the phosphate group of the phosphorylated residue, occur within five conserved sequence motifs found in other enzymes, including the purple acid phosphatase, whose common function is to catalyze phosphoryl transfer reactions to water [12]. These observations suggest that PPP and purple acid phosphatases evolved by divergent evolution from an ancestral metallophosphoesterase. Consistent with roles in catalysis, mutation of these residues either eliminates or profoundly reduces catalytic activity. PPPs catalyze dephosphorylation in a single step with a metal-activated water molecule or hydroxide ion. The most convincing evidence for this notion is that the purple acid phosphatase, which is generally related in structure to the PPPs both at the tertiary level and at the catalytic site [13], promote dephosphorylation with inversion of configuration of the oxygen geometry of the phosphate ion [14]. This indicates that a phosphoryl-enzyme intermediate would not occur. The two metal-bound water molecules are within van der Waals distance of the phosphorous atom of the phosphate bound to the catalytic site, and one of them is likely to be metal-activated nucleophile. Chapter | 86 The Structure and Topology of Protein Serine/Threonine Phosphatases Interactions with Regulatory Subunits PP1 and PP2A are responsible for regulating diverse cellular functions by dephosphorylating multiple and varied protein substrates. This seemingly paradoxical situation was resolved by the discovery that distinct forms of PP1 and PP2A holoenzymes occur in vivo, where essentially the same catalytic subunit is complexed to different targeting and regulatory subunits. For PP1 it has been shown that targeting subunits confer substrate specificity by directing particular PP1 holoenzymes to a subcellular location and by enhancing or suppressing activity toward different substrates. The control of PP1 holoenzyme structure and activity by the combinatorial selection of different targeting/regulatory subunits has recently been the subject of numerous reviews [15, 16] and will not be discussed at length here, although the contrasting structural mechanism by which the conserved PP1 and PP2A catalytic subunits are able to form diverse holoenzyme structures will be discussed. In the case of PP1, it is known that the binding of targeting subunits to its catalytic subunit is mutually exclusive, suggesting that there are one or more common or overlapping binding sites, recognized by all PP1-binding subunits. It is therefore a little surprising that PP1-binding subunits are highly diverse structurally and share little to no overall sequence similarities. The key to understanding this paradox came from the crystal structure of PP1c in complex with a short, 13-residue peptide derived from a region of the PP1-glycogen targeting subunit (GM) responsible for PP1c interactions (Figure 86.1a). This structure showed that the peptide associated with the phosphatase via two hydrophobic residues (Val and Phe), which engage a hydrophobic groove on the protein surface formed from the interface of the two -sheets of the central -sandwich and remote from the catalytic site [5]. Two basic residues of the peptide immediately N-terminal to the Val residue form salt-bridge interactions with Asp and Glu residues at one end of the peptide binding channel. Alanine substitutions of either the Val or Phe residues of the peptide abolish PP1–peptide interactions. Analysis of other PP1-binding subunit sequences revealed the presence of the identical or related sequence motif RRVxF, found to mediate the interactions between the GM peptide and PP1c [5]. The role of the degenerative RVxF motif in mediating PP1-regulatory subunit interactions is now supported by numerous experimental observations. First, for various regulatory subunits, mutation of either hydrophobic residue of the motif in native proteins weakens or eliminates their association with PP1c. Second, peptides corresponding to the RVxF motif competitively disrupt the interactions of regulatory subunits with PP1c. Third, the use of a common or overlapping PP1c binding site explains why the interactions of regulatory subunits is mutually exclusive. The number of PP1 holoenzyme structures that could be generated in this 679 manner is potentially infinite, and to date over a hundred PP1 regulatory subunits have been characterized. The residues of PP1 that interact with the RVxF peptide are conserved in all isoforms of PP1 in all eukaryotic species, although not within PP2A and PP2B, explaining why PP1-binding subunits are unique to PP1. The mechanism of combinatorial control of PP2A holoenzyme structure is different from that of PP1 and is mediated by a scaffolding subunit termed the PR65/A subunit, which simultaneously associates with the PP2A catalytic subunit and a variable regulatory B subunit. The ability to recognize a variety of regulatory subunits (perhaps over 50) is conferred by the architecture of the PR65/A subunit, which consists of 15 tandem repeats of a 39 amino-acid sequence termed the HEAT motif and related in structure to ARM repeats. These repeats assemble to create an extended molecule ideally suited for mediating protein–protein interactions [8]. Combinatorial generation of variable PP2A holoenzymes is achieved by the ability of different combinations of HEAT motifs to select different regulatory B subunits [8]. Interactions of Natural Toxins with PP1 PP1 and PP2A are specifically and potently inhibited by a variety of naturally occurring toxins such as okadaic acid, a diarrhetic shellfish poison and powerful tumor promoter, and microcystin, a liver toxin produced by blue-green algae [17]. Whereas PP2B is only poorly inhibited by the toxins that affect PP1 and PP2A, it is known to be the immunosuppressive target of FK506 and cyclosporin in association with their major cellular binding proteins, the cis-trans peptidyl prolyl isomerase FKBP12 and cyclophilin, respectively [18]. The mechanism of inhibition of PP1 by okadaic acid and microcystin LR have been defined by structures of PP1 in complex with these inhibitors. Both inhibitors, although structurally different, bind to a similar region of the phosphatase, occupying the catalytic channel to directly block phosphatase–substrate interactions. Regions of PP1 that contact the toxins include the hydrophobic groove and the 12/13 loop, the latter undergoing conformational changes to optimize contacts with microcystin [2, 3]. Both toxins disrupt substrate–phosphatase interactions by competing for sites on the protein that coordinate the phosphate group of the substrate. For example, carboxylate and carbonyl groups of microcystin interact with two of the metal-bound water molecules [2], whereas okadaic acid contacts the two phosphate-coordinating arginine residues (Arg-96 and Arg221) [3]. A similar mechanism of phosphatase inhibition by steric hindrance of substrate binding is observed in the structure of the full-length PP2B holoenzyme, for which the autoinhibitory domain lies over the substrate binding channel of the catalytic domain in such a way that a Glu side chain accepts a hydrogen bond from two of the metal-bound water molecules [6]. 680 Protein serine/threonine phosphatases of the PPM family Protein phosphatases of the PPM family are present in both eukaryotes and prokaryotes for which the defining member is PP2C. Biochemically, the PPM family was distinguished from the PPP family by its requirement for divalent metal ions (Mg2) for catalytic activity, although it is now known from crystal structures that both PPP and PPM phosphatases catalyze dephosphorylation reactions by means of a binuclear divalent metal center. Within the PPM family, the PP2C domain occurs in numerous structural contexts that reflect structural diversity [19]. For example, the PP2C domain of the Arabidopsis ABI1 gene is fused with EF hand motifs, whereas in KAPP-1, a kinase interaction domain associated with a phosphorylated receptor precedes the phosphatase domain. Other less closely related examples include the Ca2 stimulated mitochondrial pyruvate dehydrogenase phosphatase, which contains a catalytic subunit sharing 22 percent sequence identity with that of mammalian PP2C, and the SpoIIE phosphatase of Bacillus subtilus, which has ten membrane-spanning regions preceding the PP2C-like catalytic domain. A surprising homolog is a 300-residue region of yeast adenylyl cyclase present immediately N-terminus to the cyclase catalytic domain that shares sequence similarity with PP2C. This domain may function to mediate Ras-GTP activation of adenylyl cyclase activity and is not known to possess protein phosphatase activity. In eukaryotes, the various isoforms of PP2C have been implicated in diverse functions such as regulation of cell-cycle progression mediated by dephosphorylation of CDKs [20], to regulation of RNA splicing [21], control of p53 activity [22], and regulation of stress response pathways in yeast [23]. The sequences of protein phosphatases of the PPM family share no similarity with those of the PPP family, and the natural toxins that inhibit the PPP family have no affect on PPM family phosphatases. It therefore came as a surprise when the crystal structure of human PP2C revealed a striking similarity in tertiary structure and catalytic site architecture to the PPP protein phosphatases (Figure 86.1b) [19]. Mammalian PP2C consists of two domains; an Nterminal catalytic domain common to all members of the PP2C family is fused to a 90-residue C-terminal domain, unique to the mammalian PP2Cs [19]. The catalytic domain is dominated by a central, buried -sandwich of 11 -strands formed by the association of two antiparallel -sheets, both of which are flanked by a pair of antiparallel -helices inserted between the two central -strands, with four additional -helices. The C-terminal domain is formed from three antiparallel -helices remote from the catalytic site, suggesting a role in defining substrate specificity rather than catalysis. At the catalytic site of PP2C, two Mn2 ions, separated by 4 Å, form a binuclear metal center and are coordinated PART | II Transmission: Effectors and Cytosolic Events by four invariant aspartate residues and a non-conserved Glu residue (Figure 86.2b) [19]. These residues are situated at the top of the central -sandwich that forms a shallow channel suitable for the dephosphorylation of phosphoserine- and phosphothreonine-containing proteins. Six water molecules coordinate the two metal ions. One of these water molecules bridges the two metal ions and four form hydrogen bonds to a phosphate ion at the catalytic site. Dephosphorylation is probably catalyzed by a metal-activated water molecule that acts as nucleophile in a mechanism similar to that proposed for the PPP family. A recent kinetic analysis of PP2C by Denu and colleagues [24] indicated that Mn2 and Fe2 are the most effective divalent metal ions in promoting dephosphorylation reactions, suggesting that at least one of these ions must be present at the catalytic site. In contrast, Zn2 and Ca2 competitively inhibit PP2C Mn2-dependent activity. An Fe2-containing catalytic site would be analogous to the PPP protein phosphatases and possibly explains the H2O2-mediated inactivation of PP2C [25] consequent on the redox sensitivity of Fe2 and its oxidation to the inactive Fe3 valence state [24]. The finding that an ionizable group with a pKa of 7.0 has to be deprotonated for catalysis was fully consistent with the notion from the crystal structure that a metal activated water molecule acts as a nucleophile [19, 24]. Fluoride has long been used as an inhibitor of both PPP and PPM phosphatases, and the rationale for this inhibition is now clear from the catalytic mechanism of serine/ threonine phosphatases revealed by the crystal structures. By substituting for the metal-bound nucleophilic water molecule, fluoride prevents metal-activated nucleophilic attack on the phospho-protein substrate. Substitution of Ala for the Asp residues of the catalytic site in yeast and plant PP2C homologs and in the related phosphatase SpoIIE from B. subtilus abolishes catalytic activity, supporting a role for the metal ions in catalysis [26, 27]. Conclusions Although the PPP and PPM phosphatases share a similar tertiary structure, characterized by a central -sandwich and flanking -helices, with related catalytic site architectures and mechanisms, two observations suggest that these protein families have evolved from distinct ancestors. First, the secondary structure topology of the PPP and PPM families are unrelated and there is no simple rearrangement of chain connectivities that would allow the PPP topology to be transformed into the PPM topology. Second, the conserved sequence motifs of the PPP family required for metal binding and catalysis, typical of some metallophosphoesterases, are distinct from the conserved sequence motifs of the PPM family. The comparison of the PPP and PPM families of Ser/Thr protein phosphatases provides interesting contrasts with the protein phosphatases of the C(X)5R-motif superfamily composed of three distinct gene Chapter | 86 The Structure and Topology of Protein Serine/Threonine Phosphatases families: (1) the conventional protein tyrosine phosphatases (PTPs), including dual-specificity phosphatases and PTEN lipid phosphatases; (2) low-molecular-weight protein tyrosine phosphatases (lmPTPs); and (3) Cdc25 dual-specificity phosphatases [28]. These proteins share a similar overall tertiary structure composed of a central -sheet surrounded on both sides by -helices which results in an identical catalytic site architecture in all three families characterized by the nucleophilic Cys-residue located within a phosphate binding cradle. What distinguishes the three protein families are differences in their secondary structure topology; however, the topologies of the PTP and lmPTP families are related by a simple permutation of secondary structure connectivity, suggesting that these families may have evolved from the same ancestor and diverged as a result of exon shuffling events. In recent years, much has been learned of the structural details of Ser/Thr protein phosphatases relating to their overall folds, catalytic mechanisms, and interactions with regulatory subunits and toxins. However, still elusive is the structure of a Ser/Thr protein phosphatase in complex with a phosphoprotein substrate that would explain the basis for the selectivity for Ser/Thr residues and reveal the mechanism of substrate selectivity by regulatory subunits. It is hoped that future studies of Ser/Thr phosphatase will address these questions. References 1. Hubbard MJ, Cohen P. On target with a new mechanism for the regulation of protein phosphorylation. Trends Biochem Sci 1993;18:172–7. 2. Goldberg J, Huang HB, Kwon YG, Greengard P, Nairn AC, Kuriyan J. Three-dimensional structure of the catalytic subunit of protein serine/threonine phosphatase-1. Nature 1995;376:745–53. 3. Maynes JT, Bateman KS, Cherney MM, Das AK, Luu HA, Holmes CF, James MN. Crystal structure of the tumor-promoter okadaic acid bound to protein phosphatase-1. J Biol Chem 2001;276:44,078–44,082. 4. Egloff MP, Cohen PT, Reinemer P, Barford D. Crystal structure of the catalytic subunit of human protein phosphatase 1 and its complex with tungstate. J Mol Biol 1995;254:942–59. 5. Egloff MP, Johnson DF, Moorhead G, Cohen PT, Cohen P, Barford D. Structural basis for the recognition of regulatory subunits by the catalytic subunit of protein phosphatase 1. EMBO J 1997;16:1876–87. 6. Kissinger CR, Parge HE, Knighton DR, Lewis CT, Pelletier LA, Tempczyk A, Kalish VJ, Tucker KD, Showalter RE, Moomaw EW, et al. Crystal structures of human calcineurin and the human FKBP12– FK506–calcineurin complex. Nature 1995;378:641–4. 7. Griffith JP, Kim JL, Kim EE, Sintchak MD, Thomson JA, Fitzgibbon MJ, Fleming MA, Caron PR, Hsiao K, Navia MA. X-ray structure of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12–FK506 complex. Cell 1995;82:507–22. 8. Groves MR, Hanlon N, Turowski P, Hemmings BA, Barford D. The structure of the protein phosphatase 2 A PR65/A subunit reveals 681 the conformation of its 15 tandemly repeated HEAT motifs. Cell 1999;96:99–110. 9. Das AK, Cohen PW, Barford D. The structure of the tetratricopeptide repeats of protein phosphatase 5: implications for TPR-mediated protein–protein interactions. EMBO J 1998;17:1192–9. 10. Wang X, Culotta VC, Klee CB. Superoxide dismutase protects calcineurin from inactivation. Nature 1996;383:434–7. 11. Yu L, Haddy A, Rusnak F. Evidence that calcineurin accommodates an active site binuclear metal center. J Am Chem Soc 1995;117:10,147–10,148. 12. Lohse DL, Denu JM, Dixon JE. Insights derived from the structures of the Ser/Thr phosphatases calcineurin and protein phosphatase 1. Structure 1995;3:987–90. 13. Klabunde T, Strater N, Frohlich R, Witzel H, Krebs B. Mechanism of Fe(III)–Zn(II) purple acid phosphatase based on crystal structures. J Mol Biol 1996;259:737–48. 14. Mueller EG, Crowder MW, Averill BA, Knowles JR. Purple acid phosphatase: a diron enzyme that catalyses a direct phosphogroup transfer to water. J Am Chem Soc 1993;115:2974–5. 15. Bollen M. Combinatorial control of protein phosphatase-1. Trends Biochem Sci 2001;26:426–31. 16. Cohen PT. Protein phosphatase 1: targeted in many directions. J Cell Sci 2002;115:241–56. 17. MacKintosh C, MacKintosh RW. Inhibitors of protein kinases and phosphatases. Trends Biochem Sci 1994;19:444–8. 18. Liu Jr. J, Farmer JD, Lane WS, Friedman J, Weissman I, Schreiber SL Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP–FK506 complexes. Cell 1991;66:807–15. 19. Das AK, Helps NR, Cohen PTW, Barford D. Crystal structure of human protein serine/threonine phosphatase 2C at 2.0-Å resolution. EMBO J 1996;15:6798–809. 20. Cheng A, Ross KE, Kaldis P, Solomon MJ. Dephosphorylation of cyclin-dependent kinases by type 2C protein phosphatases. Genes Dev 1999;13:2946–57. 21. Murray MV, Kobayashi R, Krainer AR. The type 2C Ser/Thr phosphatase PP2C is a pre-mRNA splicing factor. Genes Dev 1999;13:87–97. 22. Takekawa M, Adachi M, Nakahata A, Nakayama I, Itoh F, Tsukuda H, Taya Y, Imai K. p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J 2000;19:6517–26. 23. Shiozaki K, Russell P. Counteractive roles of protein phosphatase 2C (PP2C) and a MAP kinase kinase homolog in the osmoregulation of fission yeast. EMBO J 1995;14:492–502. 24. Fjeld CC, Denu JM. Kinetic analysis of human serine/threonine protein phosphatase 2C. J Biol Chem 1999;274:20,336–20,343. 25. Meinhard M, Grill E. Hydrogen peroxide is a regulator of ABI1, a protein phosphatase 2C from Arabidopsis. FEBS Letts 2001;508:443–6. 26. Adler E, Donella-Deana A, Arigoni F, Pinna LA, Stragler P. Structural relationship between a bacterial developmental protein and eukaryotic PP2C protein phosphatases. Mol Microbiol 1997;23:57–62. 27. Sheen J. Mutational analysis of protein phosphatase 2C involved in abscisic acid signal transduction in higher plants. Proc Natl Acad Sci USA 1998;95:975–80. 28. Barford D, Das AK, Egloff M-P. The structure and mechanism of protein phosphatases. Insights into catalysis and regulation. Annu Rev Biophys Biomol Struct 1998;27:133–64.