Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Cell culture wikipedia , lookup
Cell-penetrating peptide wikipedia , lookup
Cre-Lox recombination wikipedia , lookup
Gene regulatory network wikipedia , lookup
Transcriptional regulation wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Silencer (genetics) wikipedia , lookup
Lipid signaling wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Two-hybrid screening wikipedia , lookup
Mitogen-activated protein kinase wikipedia , lookup
Clinical neurochemistry wikipedia , lookup
Secreted frizzled-related protein 1 wikipedia , lookup
Biochemical cascade wikipedia , lookup
Point mutation wikipedia , lookup
Endogenous retrovirus wikipedia , lookup
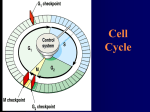
INTRODUCTION - proliferation of cells in multicellular organisms is regulated with a fine balance of stimulatory and inhibitory signals; defects in this mechanism that mediate this balance can cause cancer, which uncontrolled growth induced be either loss of function in inhibitory signals (tumor suppressors) or gain of function in stimulatory signals (oncogenes) REGULATION OF THE EUKARYOTIC LIFE CYCLE - mitosis (M phase) and DNA synthesis (S phase) are conspicuous distinct and identifiable events occur in nucleus during each of these phases - S follows M phase by G1 phase and M follows S by G2 - prior to S phase cells are diploid; after S phase cells are tetraploid; all non-cycling cells in humans are diploid; physiological growth arrest occurs during G1 phase - entry into each phase regulated by cell cycle checkpoints; cell will arrest if checkpoint requirements not met; most stringent checkpoints are prior to S and M phases Cell cycle checkpoints - G1 checkpoint: ensures cell is prepared to complete a round of DNA synthesis. - Most physiological growth arrest occurs here, as most cells in the body are 2n. - G2 checkpoint: ensures cell is prepared to undergo mitosis requires microtubules - Checkpoints are associated with cyclins. Cyclins and Cyclin-Dependent Kinases (CDKs) - progression through phases of cell cycle associated with increased expression of cyclins synthesized de novo in each cell cycle phase and degraded by proteosome following phase completion (ubiquitin mediated degradation) - specific cyclins associated with specific phases; cyclin D1 associated with G1 phase, cyclin A with S phase, cyclin B with G2/M phase, cyclin E with G1/S boundary - cyclins exists a part of multi-protein complexes that contain cyclin-dependent kinases (CDKs); specific CDKs associate with specific cyclins, such as CDK-2 with cyclin A/E, CDK-4 with cyclin D1; CDKs phosphorylate specific protein substrates on serine or threonine residues, which regulates substrate activities - resting cells (non-cycling) are diploid; most important point in cell cycle regulation occurs prior to initiation of S phase - if a cell enters S phase it is committed to divide; this point is called START and is defined as the committed point after which DNA synthesis will ensure and cell will divide - GFs removed from cells cell cycle arrests at a restriction (R point), prior to START - to traverse R-point and proceed to S phase G1 complex must be formed which contains CDK-4, PCNA, and Cyclin D; complex is active when CDK-4 phosphorylates protein pRb (tightly bound to a TF called E2F); once phosphorylated, prb-E2F complex dissociates and E2F activates set of genes needed for DNA replication (DNA polymerase); genes are START genes - pRb is an inhibitor of cell cycle tumor suppressor gene; pRb defect incidence of retinoblastoma at early age; tumor suppressor mutations are loss of function mutations - other tumor suppressor (inhibitor of cell cycle) p21WAF associates/inhibits kinase activity of complex Regulation of the cyclin D/CDK4 checkpoint - can be activated by growth promoting signals such as epidermal growth factor (EGF) or inhibited by negative growth modulators, such as TGF-beta, p27, and p15 by DNA damage via p53 induction of p21WAF or by E2F itself (which negatively feedbacks via activation of p16_ - p53 (master switch tumor suppressor gene); it is normally unstable but stabilized by ATM (protein responsible for ataxia telangiectasia) - ATM is a kinase activated by DNA damage (caused by ionizing radiation) - p53 induces p21WAF (cell cycle arrest); it also stimulates GADD-45 (growth arrest and DNA damage inducible protein-45) initiates DNA repair enzymes; p53 sense DNA damage arrests cell cycle induces DNA repair - lose p53 function accumulation of DNA mutations (cancers); p53 mutated in 60% of all human cancers; any mutated p53 inhibits holoenzyme in dominant negative fashion - p53 continuously synthesized/degraded; degradation accomplished by a ubiquitin ligase, mdm-2 adds multiple copes of ubiquitin onto p53 polyubiqiunated p53 targeted for degradation by proteosome GROWTH FACTORS AND SIGNAL TRANSDUCTION - cells require growth factor hormones to proliferate; absence of GF normal cells arrest at R-point of cell cycle; add GF (EGF) DNA replication Growth factor-receptor interaction - peptide GF bind to receptors which contain tyrosine kinase domains; these kinase domains autophosphorylate tyrosine residues on the cytoplasmic side of receptor induce specific binding of proteins which contain a src-homology region (SH2); SH2 domains have high affinity for phosphotyrosines phosphorylated cytoplasmic tail of GF receptor serves as anchor to which SH2-containing proteins bind/interact The ras and raf signaling pathway - Shc protein contains both SH2 and phosphotyrosine domains and docks GRB-2 to phosphotyrosine-containing receptor - GRB-2 (growth factor receptor binding protein) is a SH2-containing protein which activates SOS, a guanine nucleotide exchange protein which activates ras; ras is a small G-protein that is activated by binding of GTP; ras activates raf (cytoplasmic ser/thr kinase) activates the MAP kinase cascade - intrinsic GTPase activity inactivates ras by hydrolyzing GTP GDP; inhibition of activity leads to carcinogenesis Map kinase cascade - MAP (mitogen activated protein) kinases are a series of serine-threonine kinases that phosphorylate one another in a cascade amplifies signals and provides a point of integration because these kinases can be activated by signals other than the EGF signaling event; at end MAP kinase phosphorylates nuclear TFs (Elk-1, Jun) induce proliferation-associated gene transcription (START) GROWTH FACTOR FAMILIES - distinguished by structure, receptor structure, and cells specificity Class IA: EGF – Epidermal Growth Factors* - major forms are 6 kD - stimulates proliferation in epithelial, mesenchymal, and glial cells Class IB: GF – fibroblast growth factors (FGF) - acidic (16 kD) and basic (17 kD); FGF proteins are found associated with ECM and stimulate proliferation of endothelial, epithelia, mesenchymal and neuronal cells Class II: IGF- insulin-like growth factors* - 7 kD; include IGF-1 and IGF-2; related to proinsulin structurally will activate insulin receptor at high concentrations - IGf-1 produced in liver; IGF-2 produced by tumor cells; both present in plasma in conjunction with specific binding proteins - IGF stimulate glucose metabolism; they also stimulate proliferation and inhibit apoptosis Class IIIA: PDGF – Platelet-derived growth factors* - mitogenic GFs occurs as dimers of A (17 kD) and B (16 kD) chains - heterodimers (AB) and homodimers (AA, BB) exist - PDGF is derived from platelets and endothelium in adult tissues and from placenta - PDGF stimulates proliferation in mesenchymal, glial, and smooth muscle cells Class IIIB: Cytokinase/GH - interleukins (IL1-IL6), erythropoietin, and growth hormone are not GFs - they are classed as cytokines and regulate differentiation - exists as homo/hetero dimers; activate receptor that does not have intrinsic tyrosine kinase activity GROWTH FACTOR RECEPTORS - growth factor receptors contain tyrosine kinase activities and are known as receptor tyrosine kinases (RTK); distinct from type IIIB Janus-associated tyrosine kinase receptors (JAK) recruit tyrosine kinases upon activation - RTKs are specific for polypeptide growth factor hormones work in autocrine or paracrine fashion stimulate local proliferation in target tissues - three families of RTKs: I, II, and III; all activated by dimerization; Types I (EGFR) and III (PDGFR) are monomers in native (unbound) forms; types II (IGFR) is a dimer in native form Classes of RTKs - 3 classes of RTKs EGF-R, IGF-R, and PDGF-R - types I/II have one TK domain per chain; type IIIA has two TK domains per chain - type IIIA immunoglobulin-like repeats - receptor class II (insulin receptors) are dimers in native state; the ligand binding domain of the class II receptors is not imbedded in membrane, but is attached to the transmembrane subunit through a disulfide linkage - other two classes exist as monomers in their native form and are activated by ligand-induced dimerizaiton Dimerization - essential for receptor activation results in cross-phosphorylation by adjacent chains - overexpressed mutant receptors inhibit native receptors in dominant negative fashion - class I/III receptors are phosphorylated and the phosphotyrosines provide binding sites for SH2 domain-containing proteins - class II RTKs IRS-1 (insulin receptor substrate) is poly-phosphorylated to provide SH2 binding sites ONCOGENES - tumorigenic transformation caused by either viral or chemical agents - theory: cells contain a set of genes called proto-oncogenes involved in regulation of normal proliferation; chemical carcinogens disrupts the regulation of these genes/gene products cancer causing oncogene results - other proposal: viral gene products are either homologous to oncogenes or disrupt normal proto-oncogene function (viral oncogene src gene for sarcoma virus, but no cellular homolog to this viral gene product found) - cellular homolog to avian MC29 myelocytomatosis virus myc oncogene found - proto-oncogenes usually involved in proliferation regulation; most identified by homology to viral oncogene products - 6 classes of oncogenes (based on activity); proto-oncogenes themselves are not transforming and essential for normal growth Classes of oncogenes - class 1/2 growth factors and their receptors; function in growth control Class 1 oncogenes – growth factors - sis, TGF-alpha (EGF); TGF-alpha is secreted by many cell types (autocrine GF); sis secreted by sarcomas growth of sis-secreting sarcomas in vivo is suppressed by neutralizing antibodies against the sis protein showing that secretion/activity of this oncogene is necessary necessary/sufficient for unregulated growth Class 2 oncogenes – growth factor receptors - erbyB/her2-neu oncogene is an EGF receptor; when activated through overexpression this oncogene is expressed in cancers (breast) constitutively active and do not require GFs Class 3 oncogenes – non-receptor (cytoplasmic) tyrosine kinases - bcr-abl (created by chromosomal translocation) moves abl gene adjacent to an immunoglobulin promoter - immunoglobulin producing B-cells overexpress abl; immunoglobulin producing B-cells overexpress abl and lose their growth control B-cell leukemia (chronic myelogenous leukemia CML) results; abl inhibited by Gleevec Class 4 oncogenes – small G-proteins - small G-proteins prevalent in oncogenesis; ras is common mutated in 60% of cancers; it shares with p53 the distinction of being the most commonly observed mutation in cancer; two major classes of oncogenic ras proteins identified by homology to Harvey (H-ras) and Kirsten (K-ras) sarcoma viruses - mutation occurs at a specific sites which inactivates GTP hydrolyzing activity protein then becomes constitutively active; ras activates downstream MAP kinase cascade through raf Class 5 oncogenes – cytoplasmic serine/threonine kinases - c-raf initiates the MAP kinase cascade; mitogenic stimulation by GFs increase in phosphorylation of c-raf increase in c-raf activity - oncogenic forms of raf have lost N-terminal sequences that are regulatory leading to permanent activation and altered localization Class 6 oncogenes – nuclear transcription factors - nuclear proto-oncogenes are TFs which bind and activate specific promoter elements in DNA to induce transcription of specific families of gene products - in oncogenic forms these factors lose regulation or activate transcription of abnormal genes - myc is a nuclear proto-oncogene; V-myc is the transforming agents in MC29 avian myelocytomatosis virus - mitogenic agents induce expression of c-myc in most cell lines - myc has homology to other TFs with helix-loop-helix and leucine zipper motifs - myc dimerizes with protein max prior to DNA binding and myc is involved in regulating apoptosis TUMOR SUPPRESSOR GENES - mammalian cell growth is balance between growth promoting signals and growth inhibiting signals - growth promoting signals GF signal transduction; mutation result in gain-of-function (constitutively active) mutation (in proto-oncogene) and gives rise to oncogenes cancer - growth inhibitory signals observed at cell cycle checkpoints ; loss-of-function mutations in these signals result in reduced tumor suppressor activity and cancer - loss-of-function more common than gain-of-function -patient has defect in one tumor suppressor allele loss of 2nd allele occurs with high frequency complete loss of activity loss of heterozygosity (LOH) prominent mechanism in tumor suppressor mediated tumorigenesis - greater loss-of-function prevalence number/type of tumor suppressor mutations is broader than those for oncogenes - any gene that promotes well-regulated growth is a tumor suppressor - major tumor suppressor is p53 designed to protect cells against DNA double strand breaks (UV or oxidation caused); it is a TF that arrests cell cycle and induces DNA repair - if DNA damage not repaired in a given amount of time p53 can induce apoptosis and caspase activity