Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Cell growth wikipedia , lookup
Cytokinesis wikipedia , lookup
Extracellular matrix wikipedia , lookup
Tissue engineering wikipedia , lookup
Cell culture wikipedia , lookup
Endomembrane system wikipedia , lookup
Cell encapsulation wikipedia , lookup
Cellular differentiation wikipedia , lookup
Organ-on-a-chip wikipedia , lookup
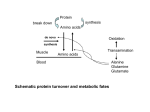
Hypothesis Glutamine breakdown in rapidly dividing cells: waste or investment? J. Carlos Aledo Summary Tumours, and in general rapidly dividing cells, behave as dissipative devices that apparently waste glutamine, since its consumption seems to exceed both energetic and nitrogen needs. Although not conclusive, there is compelling evidence suggesting that the consumption of such large amounts of glutamine is essential to sustain high rates of cellular proliferation. Herein, I first review the experimental evidence linking proliferation with high rates of glutamine breakdown. Then, the current knowledge on the proteins and activities involved in this high glutamine consumption will be summarized. Finally, the significance of the apparent waste of glutamine will be discussed on bioenergetic grounds. The discussion leads to the hypothesis that glutamine breakdown might energize some endergonic processes, as well as accelerating other exergonic processes related to cellular proliferation. BioEssays 26:778–785, 2004. ß 2004 Wiley Periodicals, Inc. Introduction Growing cells need a constant and fast supply of both energy and nitrogen substrates. Glutamine is the most abundant amino acid in the plasma, where it functions as a non-toxic nitrogen vehicle(1) and a respiratory fuel.(2) Thus, it is not surprising that glutamine behaves as a key nutrient for rapidly dividing cells. What may be surprising is the very high rate of glutamine utilization exhibited by these cells. It is widely accepted that the breakdown of glutamine greatly exceeds the anabolic and energetic requirements of proliferating cells.(3,4) In this article, I will argue that the low efficiency of glutamine metabolism can be shown to be optimal for maximal growth rate. Therefore, this apparent ‘‘waste’’ of glutamine can be explained as the cost that the organism must pay to grow rapidly. Departamento de Biologı́a Molecular y Bioquı́mica, Facultad de Ciencias, Universidad de Málaga, 29071 Málaga, Spain. E-mail: [email protected] DOI 10.1002/bies.20063 Published online in Wiley InterScience (www.interscience.wiley.com). 778 BioEssays 26.7 High rates of proliferation demand large amounts of glutamine The glutaminolytic pathway is initiated, after translocation of glutamine across the plasma and inner mitochondrial membranes,(5) by the enzyme phosphate-activated glutaminase(6) (EC 3.5.1.2), which catalyzes the hydrolysis of the amide group of glutamine, yielding stoichiometric amounts of ammonium and glutamate. Glutaminase activity correlates well with glutamine consumption and growth rate.(7) Although many cells may require large amounts of glutamine for biosynthetic purposes, only a limited amount (<5%) of the extracted glutamine is used for such pathways.(8) However, it is noteworthy that, in rapidly dividing cells, the flux through glutaminase greatly exceeds the capacity of the mitochondria to oxidize glutamate.(2) A set of detailed studies carried out in vivo has revealed a net flux of glutamine from host tissues towards tumour cells, while a reverse flux of glutamate and aspartate takes place from tumour to plasma.(1,9 –11) Interestingly, a glutamine/glutamate cycle similar to that described between tumours and their hosts appears to be operative between fetus and placenta: the placenta supplies fetus with very large amounts of glutamine, while concurrently removing glutamate from fetal circulation.(12) Thus, depending on the cell type and the metabolic requirements, a variable proportion of the glutamine-derived glutamate will be excreted as an end product, while the remaining can undergo partial oxidation to aspartate.(13) Therefore, glutamine consumption by rapidly dividing cells has been described by some authors as almost completely dissipative.(14,15) Nevertheless, there is compelling evidence pointing to this apparent waste of glutamine as an essential requisite to sustain high rates of cellular proliferation. Indeed, glutamine clearance by infusion of pure glutaminase into the bloodstream of patients suffering from cancer stops tumour growth. Unfortunately, this kind of therapy lacks specificity and interferes with other healthy rapidly dividing cells, provoking multiple side effects.(14,15) Experiments carried out using tumour cells from diverse origins, also support the view that glutamine breakdown is a process closely linked to cellular proliferation. Intracellular glutamine concentration has been negatively correlated with growth rate.(16) In contrast, high extracellular glutamine levels BioEssays 26:778–785, ß 2004 Wiley Periodicals, Inc. Hypothesis stimulate proliferation, while a reduction of glutamine availability induces phenotypical and functional differentiation.(17,18) Taking all these observations together, it seems that the key fact is glutamine hydrolysis itself. Recently, we have provided more direct evidence supporting this view. Using an antisense approach, glutaminase activity of tumour cells was partially reduced. This reduction in glutaminolytic capacity was followed by a longer doubling time and a decrease in the saturation density and plating efficiency of the transfected cells.(19) All these changes point to a moredifferentiated and less-transformed phenotype of those cells with a reduced glutaminolytic capacity. A closer look to the glutaminolytic pathway At a first glance, it is difficult to explain the glutamine cycle established between rapidly growing tumour cells and their host. The dividing cell takes up glutamine, hydrolyzes the amide group and gets rid of the products without any apparent profit. However, this is an oversimplification of the glutaminolytic pathway, which we will now look at in more detail. Glutamine uptake In mammalian cells, glutamine can be recognized as a substrate by several transport systems (Table 1A). Tumour cells exhibit enhanced transport of glutamine across their plasma membrane.(20,21) While increased amino acid trans- port could in principle be accommodated by increased expression of those transport proteins already present in the non-proliferating cells, there is evidence suggesting that the transformed phenotype involves a change in the pattern of the transporters expressed.(22) Work carried out in different laboratories, with cells of diverse origins, have led to the hypothesis that the ASC and L systems may be related to the growth and proliferation of both tumour and normal cells during tissue development.(23–32) In fact, proteins belonging to these transport agencies have been proposed as prognostic markers.(25,29) It is noteworthy that these two systems do not depend directly on the sodium electrochemical potential to drive the glutamine uptake. The possible functional significance of this fact will be discussed later. Glutamine metabolism Once glutamine is inside the cell, there are two major fates for this amino acid; either, it is excreted as glutamate plus ammonium after suffering hydrolysis in a non-redox reaction catalyzed by glutaminase, or it is partially oxidized to aspartate. This partial oxidation can be coupled to the formation of 9 moles of ATP. By contrast, if glutamine were completely oxidized, up to 27 moles of ATP could be obtained. For many years (see Box 1), the high glutamine consumption and the low ATP yield observed in proliferating cells in our opinion have been misinterpreted. It has been argued that energy Table 1. Glutamine transporters System Protein A: Glutamine and anionic amino acid transporters A ATA1 ATA2 ASC/B8 ASCT2/ATB8 Gene Mechanism and properties Slc38a1 Slc38a2 Slc1a5 Naþ-dependent. Ubiquitous expression. Short chained neutral amino acid transport. Electroneutral amino acid exchanger. Ubiquitous expression. Up-regulated in proliferating cells. Electrogenic, it takes 2 Naþ and 1 Cl per amino acid. Broad specificity for neutral and cationic substrates. Restricted expression. Naþ-independent amino acid exchanger. Broad specificity for neutral and cationic substrates. Naþ-independent ubiquitously expressed exchanger for large hydrophobic amino acids. Up-regulated in proliferating cells. Electroneutral transport of Q, N, H and in some instances, S, G and A, coupled to the inward movement of 1 Naþ and of 1 Hþ in the opposite direction. Restricted expression. Electroneutral Naþ-dependent cationic-neutral amino acid exchanger. B8,þ ATB8,þ Slc6a14 b8,þ b8,þAT Slc7a9 LAT1 LAT2 SN1 SN2 Slc7a5 Slc7a8 Slc38a3 Slc38a5 yþLAT1 yþLAT2 Slc7a7 Slc7a6 ASCT2/ATB8 EAAT1/GLAST EAAT2/GLT EAAT3/EAAC EAAT4 EAAT5 xCT Slc1a5 Slc1a3 Slc1a2 Slc1a1 Slc1a6 Slc1a7 Slc7a11 ClC-3 ? Others ? Clcn3 — L SN YþL B. Anionic amino acid transporters ASC/B8 X-AG x-c VSOAC Under slightly acid conditions it can recognize Glu and Asp as substrates. 1 Glu or Asp is taken with 3 Naþ and 1 Hþ in exchange for 1 Kþ. 30–40% identity with carriers belonging to system ASC. Down-regulated in proliferating cells. Naþ-independent electroneutral Glu/Cystine exchanger. Broadly distributed. Up-regulated in proliferating cells. Volume-sensitive organic anion channels are potential source of acidic amino acids efflux. Ubiquitous expression. BioEssays 26.7 779 Hypothesis Box 1. What is the role of high rates of glutamine utilization? Indeed, the complete oxidation of glutamine yields more moles of ATP than its partial oxidation. However, the key question is what pathway does supply ATP quicker? In other words, the stoichiometry of the process must not be confused with its kinetics, as it has often happened Year Sentence 1985 ‘‘. . . energy generation per se may not be the correct explanation for high rates of glutaminolysis in these cells since oxidation is only partial.’’ ‘‘. . . if glutamine was vitally important in energy production it would be expected that more would be converted to acetyl-CoA for complete oxidation via the Krebs cycle.’’ ‘‘. . . if energy formation per se was the major reason for the high rate of glutamine utilization, why is the oxidation only partial?’’ ‘‘. . . under conditions of extended cell culture, glutamine can be fully oxidized . . . and thus may become a major oxidative fuel under these conditions.’’ 1985 1991 1999 Reference (3) (69) (70) (71) generation may not be the correct explanation for high rates of glutaminolysis in these cells, since oxidation is only partial. Growing cells use large amounts of ATP, therefore why does the cell get rid of energetically rich substrates such as glutamate and aspartate? Here we propose just the opposite: because glutamine oxidation is only partial, ATP can be supplied at a fast enough rate for rapid proliferation. This interpretation has been hampered by confusing ATP stoichiometry (moles of ATP formed by mol of glutamine consumed) with kinetics (moles of ATP formed per unit time). In other words, rather than the amount of ATP needed for cell division, the key variable is the time required to produce it, at least when the fastest growth capacity is the main goal. This argument is well illustrated by the comparison of anaerobic glycolysis with oxidative phosphorylation. Although the yield of ATP produced by mol of glucose consumed is 18–19 times higher in oxidative phosphorylation, the rate of ATP production by glycolysis can be up to 100 times faster than that of oxidative phosphorylation.(33) Nevertheless, although a partial oxidation of glutamine to aspartate and CO2 may offer an important kinetic advantage with respect to the complete oxidation, we still have to explain why a large proportion of the glutamine utilized is returned to the medium as glutamate. Glutamate and aspartate release Mammalian cells express a number of different proteins able to mediate this transport process (Table 1B). We will focus on the 780 BioEssays 26.7 activities present in rapid-dividing cell, paying special attention to those proteins that are upregulated in cancer cells specifically. Perhaps the best understood model is the astrocytederived cancer cell. Glioma cells release large amounts of glutamate,(34) which traces an excitotoxic pathway around the tumour, thus allowing for rapid tumour expansion.(35) In addition, glutamate can act as an autocrine factor on glioma cell’s AMPA receptors, promoting migration and suppressing apoptosis.(36) Glutamate release is mainly mediated by system xc.(37) Because, in most cells, this transporter is a 1:1 cystine–glutamate exchanger, it has been proposed that the physiological role of this agency is to act as a cystine transporter that uses the transmembrane gradient of glutamate as a driving force.(38) In line with that, extracellular glutamate inhibits cystine uptake while aspartate has little(39) or no effect.(40) The increased activity of system xc observed in gliomas, is accompanied by a decrease in the uptake of glutamate mediated by excitatory amino acid transporters (EAATs).(37) Interestingly, Guo et al observed that ectopically expressed EAAT2/GLT is toxic to U251 glioma cells as well as to undifferentiated primary astrocytes.(41) In cancer cells other than those derived from the brain, dicarboxylic amino acid transport is poorly characterized. Although a defective transport of glutamate/aspartate via EAAT seems to be a trait of proliferative cells,(37,41,42) the expression of diverse EAAT isoforms in some tumour cell lines has been reported.(22,43) May EAATs operate in the efflux mode in these cells? The transport cycle is reversible at all its stages; therefore, gradients of substrate concentrations will determine the direction of the transport. The [Glu]in/[Glu]out ratio found in tumours and other tissues able to set large transmembrane glutamate gradients, is between 102 and 104.(10) Nevertheless, EAATs are very concentrative transporters because they thermodynamically couple the uptake of one molecule of glutamate to the downhill movement of three Naþ and one Kþ. The concentrative capacity of EAAT can be expressed as the transmembrane glutamate concentration ratio that will be found at equilibrium, ([Glu]in/[Glu]out)eq, which is a function of the membrane potential (Cm) and the electrochemical gradients of the ions involved in the transport process. Considering Cm 70 mV and [Naþ]i/[Naþ]o 0.1, [Kþ]o/ [Kþ]i ¼ 5 102 and [Hþ]i/[Hþ]o ¼ 2.5, which are sound values for these variables in mammalian cells, then the concentrative capacity of EAAT is around 106. This ratio is far above that usually found in any cell, and it is too high to allow the reversion of glutamate uptake. However, Rossi and coworkers have convincingly proved that glutamate release after brain ischaemia is mainly due to reversed uptake.(44) Under these conditions, ATP is depleted and the cell becomes depolarized, making feasible the operation of EAATs in the efflux mode. Interestingly, tumour cells may exhibit higher (depolarized) membrane potential with respect to normal tissues. For instance, the resting membrane potentials of Hypothesis unsynchronized MCF-7 cells during exponential growth phase, when measured using sharp glass microelectrodes, range from 58.6 mV to 2.7 mV.(45) Furthermore, the mean membrane potential in breast biopsy tissue from women with infiltrating ductal carcinoma is significantly depolarized, compared with values measured in tissue from women with benign breast disease.(46,47) In this context, I feel tempted to speculate that, in certain tumours and under certain circumstances, the release of glutamate could partially contribute to the maintenance of the electrochemical gradients of sodium and potassium, by driving the transport mediated by EAAT outwards. Regardless of the validity of this speculative hypothesis, tumours overexpressing EAATs are unusual.(43) The trend seems to be just the opposite, that is, cellular transformation is accompanied by a downregulation of EAATs and an increase in the activities of systems xc and ASC/ B0.(13,20,48) System ASC was originally described by Christensen as an entity serving for zwitterionic amino acid transport.(49) Later on, this author(50) and others(51) realized that the same agency can also serve as a transporter for anionic amino acids under slightly acidic conditions, while the transport of glutamine by this carrier shows little pH dependence.(51) Stimulation of anionic amino acids transport via ASC by reduction of the ambient pH has been widely documented in several cellular systems. The effect of the pH has been ascribed to protonation of the anionic substrate(51) but mostly to protonation of the transporter(50) that results in an increased affinity for glutamate.(52) Although measurement of pH in vivo has shown that the microenvironment in tumours is generally more acidic than in normal tissue,(53) one might yet question the physiological importance of ASC in mediating glutamate transport. However, Nunck and Nunck have shown that, because of its high capacity, system ASC can transport glutamate at significant rates, even in the presence of serine at pH 7.2.(52) Furthermore, ASCT2 has been proposed to mediate the observed efflux of L-aspartate across the blood–brain barrier.(54) Thus, ASC could help to drive a glutamine– aspartate (or glutamate) exchange, while an important part of the glutamine-derived glutamate could be exchanged with cystine via system xc, which has been linked to proliferation in both tumour and embryonic tissues. In the intracellular environment, cystine is reduced to cysteine,(55) the ratelimiting substrate for glutathione synthesis. Glutathione levels are of critical importance to tumour cells, affecting their ability to withstand oxidative attack.(56) Furthermore, the chemosensitivity of tumour cells has been related to intracellular glutathione levels.(57) Can glutamine hydrolysis energize amino acid transport? Transport across the plasma membrane is essential for supplying cells with nutrients for cellular metabolism. Many of these nutrients are available in the extracellular milieu in lower concentrations than those required within the cell. Such substances must be actively transported. Most animal cells make use of the existing Naþ gradient as a driving force that energizes transport processes. This Naþ gradient is created and maintained with the participation of the (Naþ,-Kþ)ATPase. This protein pumps three Naþ out of the cell and two Kþ into the cell, against their respective electrochemical gradients at the expense of one molecule of ATP. The (Naþ,Kþ)-ATPase is one of the single major users of cellular energy, responsible for 5–40% of the steady-state energy consumption.(58) Rapidly dividing cells possess an enhanced metabolism accompanied by faster nutrient uptake. This means that a high amount of the cellular ATP produced should be expended in powering transport, to the detriment of other ATP-utilizing processes such as macromolecule biosynthesis, which is quite strongly controlled by ATP supply.(59) The idea that I would like to put forward is simple. If the cell were able to pay the active transport bill with a currency that did not involve ATP (neither directly nor indirectly through the generation of Naþ gradients across the membrane), it would give an important advantage. Furthermore, the established paradigm, that the Naþ gradient is sufficient to explain the steady-state entrance of amino acids in eukaryotic cells, has been questioned.(60,61) Consequently, other energy sources need to be postulated for amino acid transport. Because growth is an energetically expensive task, a maximal rate of proliferation must be paralleled by an optimal ability to withdraw energy from the environment. In order to illustrate this point, let’s think about a simplified cell model, where an energetic substrate, say glutamine, is oxidized with the concomitant production of ATP. If this catabolic pathway is working at maximal capacity, then any increase in glutamine availability will not be reflected in a further increase in ATP supply. However, if under these circumstances, the cell were able to use the excess of glutamine, not to make ATP but to drive the uptake of nutrients, this would give an obvious advantage in growing cells. In this context, I hypothesize that glutamine hydrolysis free energy can partially contribute to the energization of amino acid uptake in rapidly dividing cells. At this point, it may be convenient to summarise what I have reviewed concerning the glutaminolytic pathway in a previous section. This summary is illustrated in a schematic drawing in Fig. 1. Briefly, (1) rapidly dividing cells show high fluxes of glutamine (inwards), glutamate (outwards) and aspartate (outwards). (2) Transport systems ASC, L and xc are related to proliferation. (3) System ASC allows the interchange of neutral amino acids by aspartate or glutamate. (4) A significant amount of the glutamine-derived glutamate seems to cross the membrane via system xc, in interchange with cystine. (5) Upon cell entry cystine is rapidly reduced to cysteine.(38) (6) Tumour cells establish a large transmembrane gradient of cysteine.(10) (7) Cysteine is a good substrate for systems L and ASC, and therefore it can be interchanged for other amino acids. (8) Outside the cell, under BioEssays 26.7 781 Hypothesis Figure 1. Systems ASC, L, xc and glutaminase form the energy transduction machinery coupling the glutamine breakdown to the uptake of amino acids. A high glutaminase activity (GA) facilitates the buildup of an intracellular glutamate pool. This pool can be directly or indirectly used to drive the endergonic uptake of those amino acids recognized as substrates by systems xc, L and ASC. Glutamate can also be partially oxidized to aspartate, which not only supply ATP but also may help to speed up the glutamine uptake in the growing cell. Aa0 represents those amino acids related to glutamine metabolism, such as Ala and Cys, that are highly concentrated within the cell and they are good substrates for system ASC. an oxidant environment, cysteine is likely to be oxidized to cystine and again serves as substrate for cystine–glutamate exchange.(37) From the above exposition, it follows that a high glutaminase activity, able to exceed the capacity of the mitochondria to oxidize glutamate, facilitates the buildup of an intracellular glutamate pool. This pool could be directly used to drive the endergonic uptake of those amino acids recognized as substrates by system ASC. Nevertheless, we must note that system ASC as a substantial contributor to anionic amino acid transport is a hypothesis and not a confirmed mechanism. Indirectly, the potential energy stored in the intracellular glutamate pool could be used to speed up the uptake of cystine via system xc; this, in turn, would result in the establishment of a cysteine gradient across the cell membrane, which could drive the uphill uptake of those amino acids susceptible to being interchanged with cysteine via system L and ASC. In this sense, plasma glutamine represents a pool of chemical free energy that could be exploited by rapidly dividing cells. One may question why evolution has not led to the establishement, in every mammalian cell, of such a bioenergetic strategy. If we consider the energetic economy of the whole organism, it becomes obvious that such strategy is very costly. In order to keep the levels of plasmatic glutamine constant, organs such as liver, kidney and muscle must expend large amounts of ATP to synthesize and release 782 BioEssays 26.7 glutamine.(62) If the thermodynamic system considered now is the organism as a whole, it can be stated that the uphill amino acid uptake in the rapid dividing cells is energized by the hydrolysis of ATP which has taken place far away, in those tissues that show a net glutamine production. In this view, glutamine can be described as an energy transducing agent. Glutamine breakdown speeds up the metabolism of amino acids Taking data from the literature,(10,63,64) I have calculated the actual Gibbs free-energy change for the interchange of amino acids catalyzed by systems ASC, L and xc (Fig. 2). The DG0 calculated in this manner, can be regarded as an index of the accumulative potential of the transporter under current conditions. The interchange of sodium ions by amino acids that are substrates of system A, has been included for comparative purposes. It can be noted that the active uptake of amino acids driven by sodium ions may become seriously threatened in depolarized cells (Fig. 2, see the sign for the uptake of amino acid through system A). It is noteworthy to emphasize that this observation may be physiologically relevant because tumour cells exhibit higher (depolarized) membrane potential with respect to normal tissues. In contrast, transport processes working far from equilibrium are less sensitive to depolarization. Fig. 2 also shows the thermodynamic efficiency of each transport process. The Hypothesis Figure 2. Thermodynamic analysis of amino acid transport in growing Ehrlich tumour cells. Taking data from the literature,(10,63,64) the thermodynamic efficiencies and average actual Gibbs free-energy changes for the interchange of amino acids catalyzed by systems ASC, L and xc, have been calculated. Aa and Aa 0 represent the substrates for ASCT2/ATB8 and LAT2, respectively. The cotransport of sodium ions with Aa00 , amino acids that are substrates for system A, has been included for comparative purposes. All the calculations have been carried out at two membrane potential: 58.6 and 2.7 mV. These are reported values for tumour cells growing exponentially.(45) efficiencies were calculated using the equation: h¼ ðDGdriven =DGdriver Þ 100% ð1Þ With the exception of system A, which is operating near equilibrium, the calculated thermodynamic efficiencies are well below 100%. I would like to stress that these low efficiencies can be understood as optimal with respect to growth rate. Indeed, low thermodynamic efficiencies are related to high fluxes.(65) In the extreme situation of 100% efficiency, the system is at equilibrium and thus, the rate of the process is zero. In contrast, negative values are obtained for the efficiencies of the interchanges mediated by systems ASC and xc. Negative thermodynamic efficiency may need some clarification. Usually, the driver process provides the freeenergy that is necessary to impulse the driven process against its own free-energy. In these cases the efficiency is always a value in the range between 0 and 100%. However, when an exergonic process is coupled to another exergonic process, we obtain a negative value for the efficiency of this coupling. One may question, what is the point of coupling two exergonic processes? The answer is to speed up the global process. Although this conclusion can be formally derived from the phenomenological non-equilibrium thermodynamics,(66) herein, I will provide only an intuitive explanation. To this end, I would like to give an example previously used by Westerhoff et al.(67) Let us think about the combustion of petrol to move a car up a hill. Part of the free-energy released in petrol combustion is recovered in the form of increased gravitational potential energy of the car; this is a process with a positive thermodynamic efficiency because an exergonic (petrol combustion) and an endergonic (car moving uphill) process, are coupled. Nevertheless, the car can also operate at a negative efficiency: when going downhill, pressing the gas pedal will couple the petrol combustion (exergonic process) to the descending movement of the car (also an exergonic process), yet this can be useful because the car reaches its destination more quickly. Conclusions Glutamine can be removed from the plasma, hydrolyzed within the cell, and the products released to the plasma. The calculated value for the actual Gibbs free-energy of the global process is 49.3 kJ/mol.1 That is, the hydrolysis of one mole of glutamine can release almost as much free energy as the hydrolysis of ATP (50 kJ/mol).(68) Thus, plasma glutamine represents a pool of chemical free energy that can be released upon hydrolysis. Nevertheless, this high potential for glutamine hydrolysis cannot affect the potential for change in other systems unless some form of mechanical coupling exists between them. In the case of rapidly dividing cells, it is suggested that glutaminase and systems ASC, L and xc are key elements of the energy converter that couples the breakdown of glutamine to the uphill uptake of nutrients. Not surprisingly, all these proteins have been shown to be overexpressed in rapidly dividing cells. When the energetic economy of the whole organism is considered, it should be noted that such strategy is very costly, since non-proliferating tissues must expend large amounts of ATP to provide the glutamine consumed by the rapidly growing cells. In this sense, glutamine can be described as an energy transducing agent. In other words, glutamine allows the coupling of exergonic and endergonic processes that do not necessarily take place, either simultaneously or in the same tissue. This versatility has a cost in terms of thermodynamic efficiency. 1 Data and Calculations can be provided on request. BioEssays 26.7 783 Hypothesis It could be said that some efficiency must be sacrificed to make the process run faster. Once, François Jacob wrote ‘‘the dream of every cell is to become two cells’’. Using Jacob’s aphorism, it could be said that making this dream come true costs large amounts of ATP, but making the dream come true quickly costs large amounts of ATP and lots of glutamine. Acknowledgments The author is grateful to Alicia Esteban del Valle and Miguel Angel Medina for their comments on the manuscript. References 1. Carrascosa JM, Martı́nez P, Núñez de Castro I. 1984. Nitrogen movement between host and tumor in mice inoculated with Ehrlich ascetic tumor cells. Cancer Res 44:3831–3835. 2. Moreadith RW, Lehninger AL. 1984. The pathways of glutamate and glutamine oxidation by tumor cell mitochondria. J Biol Chem 259:6215– 6221. 3. Newsholme EA, Crabtree B, Ardawi MSM. 1985. The role of high rates of glycolysis and glutamine utilization in rapidly dividing cells. Biosci Reports 5:393–400. 4. Medina MA, Sánchez-Jiménez F, Márquez J, Quesada AR, Núñez de Castro I. 1992. Relevance of glutamine metabolism to tumor cell growth. Mol Cell Biochem 113:1–15. 5. Molina M, Segura JA, Aledo JC, Medina MA, Núñez de Castro I, et al. 1995. Glutamine transport by vesicles isolated from tumour-cell mitochondrial inner membrane. Biochem J 308:629–633. 6. Aledo JC, de Pedro E, Gómez-Fabre PM, Núñez de Castro I, Márquez J. 1997. Submitochondrial localization and membrane topography of Ehrlich ascitic tumour cell glutaminase. Biochim Biophys Acta 1323:173–184. 7. Aledo JC, Segura JA, Medina MA, Alonso FJ, Núñez de Castro I, et al. 1994. Phosphate-activated glutaminase expression during tumor development. FEBS Lett 341:39–42. 8. Curthoys NP, Watdford M. 1995. Regulation of glutaminase activity and glutamine metabolism. Annu Rev Nutr 15:133–159. 9. Quesada AR, Medina MA, Márquez J, Sánchez-Jiménez F, Núñez de Castro I. 1998. Contribution by host tissues to circulating glutamine in mice inoculated with Ehrlich ascitic tumor cells. Cancer Res 48:1551–1553. 10. Márquez J, Sánchez-Jiménez F, Medina MA, Quesada AR, Núñez de Castro I. 1989. Nitrogen metabolism in tumor bearing mice. Arch Biochem Biophys 268:667–675. 11. Márquez J, Núñez de Castro I. 1991. Mouse liver free amino acids during the development of Ehrlich ascites tumour. Cancer Lett 58:221–224. 12. Battaglia FC. 2000. Glutamine and glutamate exchange between the fetal liver and the placenta. J Nutr 130:974S–977S. 13. Collins CL, Wasa M, Souba WW, Abcouwer SF. 1998. Determinants of glutamine dependence and utilization by normal and tumor-derived breast cell line. J Cell Physiol 176:166–178. 14. Medina MA. 2001. Glutamine and cancer. J Nutr 131:2539S–2542S. 15. Souba WW. 1993. Glutamine and cancer. Ann Surg 218:715–728. 16. Sebol JS, Weber G. 1984. Negative correlations of L-glutamine concentration with proliferation rate in rat hepatomas. Life Sci 34:301– 306. 17. Spittler A, Oehler R, Goetzinger P, Holzer S, Reissner CM, et al. 1997. Low glutamine concentrations induce phenotypical and functional differentiation of U937 myelomonocytic cells. J Nutr 127:2151–2157. 18. Turowski GA, Rashid Z, Hong F, Madri JA, Basson MD. 1994. Glutamine modulates phenotype and stimulates proliferation in human colon cancer cell lines. Cancer Res 54:5974–5980. 19. Lobo C, Ruı́z-Bellido MA, Aledo JC, Márquez J, Núñez de Castro I, et al. 2000. Inhibition of glutaminase expression by antisense mRNA decreases growth and tumourigenicity of tumour cells. Biochem J 348: 257–261. 20. Bode BP, Souba WW. 1999. Glutamine transport and human hepatocellular transformation. J Parenter Enteral Nutr 23:S33–S37. 784 BioEssays 26.7 21. Bode BP. 2001. Recent molecular advances in mammalian glutamine transport. J Nutr 131:2475S–2485S 22. McGivan JD. 1998. Rat hepatoma cells express novel transport system for glutamine and glutamate to those present in normal rat hepatocytes. Biochem J 330:255–260. 23. Bode BP, Fuchs BC, Hurley BP, Conroy JL, Suetterlin JE, et al. 2002. Molecular and functional analysis of glutamine uptake in human hepatoma and liver-derived cells. Am J Physiol 283:G1062–G1073. 24. Dolinska M, Dybel A, Zablocka B, Albrecht J. 2003. Glutamine transport in C6 glioma cells shows ASCT2 system characteristics. Neurochem Int 43:501–507. 25. Witte D, Ali N, Carlson N, Younes M. 2002. Overexpression of the neutral amino acid transporter ASCT2 in human colorectal adenocarcinoma. Anticancer Res 22:2555–2557. 26. Wasa M, Wang HS, Okada A. 2002. Characterization of L-glutamine transport by a human neuroblastoma cell line. Am J Physiol 282:C1246– C1253. 27. Hara K, Kudoh H, Enomoto T, Hashimoto Y, Masuko T. 1999. Malignant transformation of NIH3T3 cells by overexpression of early lymphocyte activation antigen CD98. Biochem Biophys Res Commun 262:720– 725. 28. Yagita H, Masuko T, Hashimoto Y. 1986. Inhibition of tumor cell growth in vitro by murine monoclonal antibodies that recognize a proliferationassociated cell surface antigen system in rats and human. Cancer Res 46:1478–1484. 29. Nikolova M, Guenova M, Taskov H, Dimitrova E, Staneva M. 1998. Levels of expression of CAF7 (CD98) have prognostic significance in adult acute leukemia. Leuk Res 22:39–47. 30. Yanagida O, Kanai Y, Chairoungdua A, Kim DK, Segawa H, et al. 2001. Human L-type amino acid transporter 1 (LAT1): characterization of function and expression in tumor cell lines. Biochem Biophys Acta 1514:291–302. 31. Wolf DA, Wang S, Panzica MA, Bassily NH, Thompson NL. 1996. Expression of a highly conserved oncofetal gene, TA1/E16, in human colon carcinoma and other primary cancers: homology to Schistosoma mansoni amino acid permease and Caenorhabditis elegans gene products. Cancer Res 56:5012–5022. 32. Sang J, Lim YP, Panzica M, Finch P, Thompson NL. 1995. TA1, a highly conserved oncofetal complementary DNA from rat hepatoma, encodes an integral membrane protein associated with liver development, carcinogenesis, and cell activation. Cancer Res 55:1152–1159. 33. Voet D, Voet JG. 1995. Biochemistry, 2nd ed. New York: Wilely. p 464– 470. 34. Ye ZC, Sontheimer H. 1999. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res 59:4383–4391. 35. Takano T, Lin JHC, Arcuino G, Gao Q, Yang J, et al. 2001. Glutamate release promotes growth of malignant gliomas. Nat Med 7:1010–1015. 36. Ishiuchi S, Tsuzuki K, Yoshida Y, Yamada N, Hagimura N, et al. 2002. Blockage of Ca2þ-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat Med 8:971– 978. 37. Ye ZC, Rothstein JD, Sontheimer H. 1999. Compromised glutamate transport in human glioma cells: reduction-mislocalization of sodiumdependent glutamate transporters and enhanced activity of cystineglutamate exchange. J Neurosci 19:10767–10777. 38. Bannai S, Ishii T. 1988. A novel function of glutamine in cell culture: utilization of glutamine for the uptake of cystine in human fibroblasts. J Cell Physiol 137:360–366. 39. Sato H, Tamba M, Ishii T, Bannai S. 1999. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem 274:11455–11458. 40. Bannai S. 1986. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem 261:2256–2263. 41. Guo H, Lai L, Butchbach MER, Lin CLG. 2002. Human glioma cells and undifferentiated primary astrocytes that express aberrant EAAT2 mRNA inhibit normal EAAT2 protein expression and prevent cell death. Mol Cell Neurosci 21:546–560. 42. Mordrelle A, Jullian E, Costa C, Corment-Boyaka E, Benamouzig R, et al. 2000. EAAT1 is involved in transport of L-glutamate during differentiation of the Caco-2 cell line. Am J Physiol 279:G366–G373. Hypothesis 43. Pollard M, McGivan JD. 2000. The rat hepatoma cell line H4-II-E-C3 express high activities of the high-affinity glutamate transporter GLT-1A. FEBS Lett 484:74–76. 44. Rossi DJ, Oshima T, Attwell D. 2000. Glutamate release in severe ishaemia is mainly by reversed uptake. Nature 403:316–321. 45. Wonderlin WF, Woodfork KA, Strobl JS. 1995. Changes in membrane potential during the progression of MCF-7 human mammary tumor cells through the cell cycle. J Cell Physiol 165:177–185. 46. Marino AA, Iliev IG, Schwalke MA, Gonzalez E, Marier KC, et al. 1994. Association between cell membrane potential and breast cancer. Tumor Biol 15:82–89. 47. Cuzick J, Holland R, Barth V, Davies R, Faupel M, et al. 1998. Electropotential measurements as a new diagnostic modality for breast cancer. Lancet 352:359–363. 48. Makowske M, Christensen HN. 1982. Contrast in transport systems for anionic amino acids in hepatocytes and hepatoma cell line HTC. J Biol Chem 257:5663–5670. 49. Christensen HN, Liang M, Archer EG. 1967. A distinct Naþ-requiring transport system for alanine, serine, cysteine, and similar amino acids. J Biol Chem 242:5237–5246. 50. Vadgama JV, Christensen HN. 1984. Wide distribution of pH-dependent service of transport system ASC for both anionic and zwitterionic amino acids. J Biol Chem 259:3648–3652. 51. Utsunomiya-Tate N, Endou H, Kanai Y. 1996. Cloning and functional characterization of a system ASC-like Naþ-dependent neutral amino acid transporter. J Biol Chem 271:14883–14890. 52. Munck BG, Munck LK. 1999. Effects of pH changes on systems ASC and B in rabbit ileum. Am J Physiol 276:G173–G184. 53. Webb SD, Sherratt JA, Fish RG. 1999. Mathematical modelling of tumour acidity: regulation of intracellular pH. J theor Biol 196:237–250. 54. Tetsuka K, Takanaga H, Ohtsuki S, Hosoya K, Terasaki T. 2003. The L-isomer-selective transport of aspartic acid is mediated by ASCT2 at the blood-brain barrier. J Neurochem 87:891–901. 55. Bannai S, Ishii T. 1982. Transport of cystine and cysteine and cell growth in culture human diploid fibroblasts: effect of glutamate and homocysteate. J Cell Physiol 112:265–272. 56. Matés JM, Pérez-Gómez C, Núñez de Castro I, Asenjo M, Márquez J. 2002. Glutamine and its relationship with intracellular redox status, oxidative stress and cell proliferation/death. Int J Biochem Cell Biol 34:439–458. 57. Okuno S, Sato H, Kuriyama-Matsumura K, Tamba M, Wang H, et al. 2003. Role of cystine transport in intracellular glutathione level and 58. 59. 60. 61. 62. 63. 64. 65. 66. 67. 68. 69. 70. 71. cisplatin resistance in human ovarian cancer cell lines. Brit J Cancer 88:951–956. Ewart HE, Klip A. 1995. Hormonal regulation of the Naþ-Kþ-ATPase: mechanisms underlying rapid and sustained changes in pump activity. Am J Physiol 269:C295–C311. Buttgereit F, Brand MD. 1995. A hierarchy of ATP-consuming processes in mammalian cells. Biochem J 312:163–167. Garcı́a-Sancho J, Sánchez A, Handlogten ME, Christensen HN. 1977. Unexpected additional mode of energetization of amino-acid transport into Ehrlich cells. Proc Natl Acad Sci USA 74:1488–1491. Medina MA, Núñez de Castro I. 1990. Plasma membrane oxidoreduction and amino acid transport. In: Crane FL, Morré DJ, Löw HE, editors. Oxidoreduction at the plasma membrane: relation to growth and transport. Boca Raton: CRS Press. p. 247–255. Aledo JC, de Pedro E, Gómez-Fabre PM, Núñez de Castro I, Márquez J. 2000. Changes in mRNAs for enzymes of glutamine metabolism in the tumor-bearing mouse. Anticancer Res 20:1463–1466. de Graaf-Hess A, Trijbels F, Blom H. 1999. New method for determining cystine in leukocytes and fibroblasts. Clin Chem 45:2224–2228. Hidelbrandt W, Kinscherf R, Hauer K, Holm E, Dröge W. 2002. Plasma cystine concentration and redox state in aging and physical exercise. Mech Ageing Dev 123:1269–1281. Aledo JC, Esteban del Valle A. 2002. Glycolysis in wonderland: the importance of energy dissipation in metabolic pathways. J Chem Educ 79:1336–1339. Aledo JC. 2001. Metabolic pathways: does the actual Gibbs freeenergy change affect the flux rate? Biochem Mol Biol Educ 29:142– 143. Westerhoff HV, Hellingwerf KJ, van Dam K. 1982. Thermodynamic efficiency of microbial growth is low but optimal for maximal growth rate. Proc Natl Acad Sci USA 80:305–309. Mathews CK, van Holde KE, Ahern KG. 2002. Biochemistry, 3rded. San Francisco: Addison Wesley Longman. p. 463–498. Newsholme EA, Crabtree B, Ardawi MS. 1985. Glutamine metabolism in lymphocytes: its biochemical, physiological and clinical importance. Q J Exp Physiol 70:473–489. Newsholme EA, Board M. 1991. Application of metabolic-control logic to fuel utilization and its significance in tumor cells. Adv Enzyme Regul 31:225–246. Newsholme P, Curi R, Pithon-Curi TC, Murphy CJ, Garcia C, et al. 1999. Glutamine metabolism by lymphocytes, macrophages, and neutrophils: its importance in health and disease. J Nutr Biochem 10:316–324. BioEssays 26.7 785