Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Metabolic syndrome wikipedia , lookup
Hormone replacement therapy (female-to-male) wikipedia , lookup
Hypothalamus wikipedia , lookup
Polycystic ovary syndrome wikipedia , lookup
Growth hormone therapy wikipedia , lookup
Hormone replacement therapy (male-to-female) wikipedia , lookup
Androgen insensitivity syndrome wikipedia , lookup
Hypothalamic–pituitary–adrenal axis wikipedia , lookup
Pituitary apoplexy wikipedia , lookup
Hypopituitarism wikipedia , lookup
Hyperandrogenism wikipedia , lookup
Congenital adrenal hyperplasia due to 21-hydroxylase deficiency wikipedia , lookup
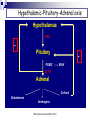
Adrenal chemistry Suki Sankaralingam Consultant Clinical Scientist MSc Essex University March 2010 Adrenal glands Adrenal glands are triangular in shape Lie superiorly and anteriorly to the kidneys Each weighs ~4g Adrenal glands Also known as suprarenal glands MSc Essex University March 2010 Adrenal glands and endocrine systems Cortex (80 -90%) Makes steroids under the influence of ACTH Medulla (10 -20%) Nervous tissue secretes catecholamines MSc Essex University March 2010 Adrenal glands and endocrine systems Adrenal glands Adrenal cortex – corticosteriod hormones Zona glomerulosa - regulated by angiotensin II Mineralocorticoids (aldosterone) Zona fasciculata – regulated by ACTH Glucocorticoids (cortisol) Zona reticularis - regulated by ACTH Androgens (DHEA, DHEAS) Adrenal medulla – chromaffin cells Adrenaline Noradrenaline MSc Essex University March 2010 Adrenal Steroids and Pathways Cholesterol 17a Pregnenolone 17-Hydroxypregnenolone 3 Progesterone 21 Deoxycorticosterone 11 Cotricosterone L Dehydroepiandrosterone 3 3 17a 17-Hydroxyprogesterone 21 Deoxycortisol Androstenedione 17ß A Testosterone Oestrone 11 Cortisol 18 Aldosterone Glucocorticoids Mineralocorticoids MSc Essex University March 2010 Androgens Enzymes in Steroid Biosynthesis Side-chain cleavage enzyme; desmolase (CYP11A1) 3 beta-hydroxysteroid dehydrogenase (3 beta HSD) 17 alpha-hydroxylase/17,20 lyase (CYP17) 21-hydroxylase (CYP21A2) 11 beta-hydroxylase (CYP11B1) 18 hydroxylase (aldosterone synthase CYP11B2) 17 beta-hydroxysteroid dehydrogenase Aromatase (CYP19) The enzyme 17α-hydroxylase (CYP 17) is not present in the outer layer of the cortex Steroids and their metabolic by-products are released into the adrenal circulation and inhibit critical enzymes in subsequent layers through which the blood flows. cortisol and androgens cannot be formed in this layer. no aldosterone can be synthesized by cells below the outer glomerulosa layer. In the inner layer - 17α-hydroxyprogesterone cannot be converted to cortisol but is shunted into the formation of androgens. Mutation or failure of any of these genes can lead endocrine disease MSc Essex University March 2010 Steroid Hormones Not encoded in genes, but derived from cholesterol through enzymatic reactions Cholesterol is converted to pregnenolone Pregnenolone moves between mitochondria and endoplasmic reticulum and is precursor to all steroids Includes Glucocorticoids, Mineralocorticoids, Androgens. MSc Essex University March 2010 Glucocorticoid receptors Receptors for glucocorticoids (GRs) are usually intracellular, exist in the cytoplasm, not the nucleus, and are associated with heat shock proteins. The site of receptor binding on the DNA is known as the glucocorticoid response element (GRE). The structural similarities of the DNA-binding domain of glucocortiocoid, oestrogen, androgen and progesterone receptors are such that they can all bind to the same hormone response element, a consensus 15 nucleotide sequence. Additionally, cortisol has equal affinity for the aldosterone receptor in the kidney tubules but its rapid inactivation to cortisone in these cells normally prevents binding. Mineralocorticoid- Aldosterone Promotes Retention of sodium Excretion of potassium and hydrogen ions mainly in distal and collecting tubes Essential hormone and 30-50 times more potent than deoxycorticosterone DNA hormone response element binds to intracellular mineralocorticoid receptor (type1) and glucocorticoid receptors (type2) by means of zinc domain Renin-Angiotensin system and circulating potassium are the most important regulators of aldosterone secretion. The effect of ACTH is short lived Hypothalamic-Pituitary-Adrenal axis Hypothalamus CRH Pituitary POMC MSH ACTH Adrenal Cortisol Aldosterone Androgens MSc Essex University March 2010 Renin –Angiotensin-Aldosterone Pathway Amino Hormones Derived from the amino acid tyrosine Includes the catecholamines: adrenaline and noradrenaline Tyrosine MSc Essex University March 2010 Adrenaline Catecholamine synthesis pathway ADR = adrenaline DA = dopamine DBH = dopamine-β-hydroxylase MAO = monoamine oxidase MHPG = 3-methoxy-4hydroxyphenylethylene glycol 3-MT = 3-methoxytyramine NM = normetadrenaline NORADR = noradrenaline PHE = phenylalanine TYR = tyrosine VMA = vanillylmandelic acid. Catecholamines Noradrenaline is formed in the adrenal medulla It is further metabolised to adrenaline The adrenaline produced and released by the adrenal gland functions as a hormone It is the elevations of one or both noradrenaline and adrenaline in the bloodstream that cause the distinctive but variable symptoms of pheochromocytoma Pheochromocytomas, similar to adrenal medullary cells, secrete catecholamines directly into the bloodstream Pheochromocytomas secrete noradrenaline whereas the predominant catecholamine secreted by the adrenal medulla is adrenaline. Actions of glucocorticoids Potent metabolic effects on many tissues Anabolic in the liver Catabolic in muscle and fat Regulates the metabolism of protein, carbohydrates and fats Diabetogenic opposing the action of insulin in peripheral tissues (decreasing glucose uptake via GLUT4 receptors) increasing glucose production and release from the liver accomplished through gluconeogenesis using amino acids (from the catabolic actions on muscle) as the primary carbon source Actions of glucocorticoids In the cardiovascular system, sustains normal blood pressure by maintaining normal myocardial function and the responsiveness of arterioles to catecholamines and angiotensin II. Inhibits the production of inflammatory factors resulting from injury. Inhibits fibroblast proliferation and the formation of collagen. Decreases osteoblast function and new bone formation Decrease gut calcium absorption and decrease renal calcium reabsorption, thus adversely affecting calcium balance. In the CNS, can alter the excitability of neurons, induce neuronal death and can affect the mood and behaviour of individuals. Cortisol secretion Secreted in a diurnal pattern Levels highest in the morning (8 -9AM) Lowest around midnight. Diurnal pattern Changes in people who work alternate shifts and sleep at different times during the day. Disrupted in people who have Cushing’s syndrome. Actions of adrenal androgens Role of DHEA and its sulphate in normal physiology – not clearly defined. DHEA, DHEAS and androstenedione - ‘weak androgens’ have a much lower affinity for the androgen receptor than testosterone. Adrenal androgens are converted peripherally to the more active testosterone. Males - physiologically insignificant compared to the amount secreted by the testes Females - adrenal-derived testosterone is important in maintaining normal pubic and axillary hair. After the menopause, adrenal androgens may also be an important source of oestradiol, again due to peripheral conversion. Adrenal androgen hypersecretion does not cause any clinical signs in adult males but is detectable in females by signs of hirsutism and masculinisation. Transport of steroids in plasma Steroid Cortisol Aldosterone Progesterone Testosterone Oestradiol % bound % bound to to Total conc. (nmol/l) % unboun d CBG Albumin SHBG t1/2in circulation (min) 400 4 90 6 0.1 100 0.4 40 20 40 0.1 10 0.6 2.4 17 80 0.6 5 20 2.0 3 40 55 10 0.1 2.0 0 68 30 20 Actions of Catecholamines Catecholamines act on their target tissues through G-protein-linked membrane receptors. Cardiovascular Increase in heart rate Increased venous return Increased peripheral resistance Visceral Smooth muscle relaxation and contraction Modulation of fluid and electrolyte transport in the gut, kidney, gall bladder Metabolic Glycogenolysis, lipolysis Water and electrolyte metabolism Decreased sodium excretion and glomerular filtration Effects on renin secretion leads to increased aldosterone production Hormone secretion Modulates the responses of a number of endocrine systems, including: The renin-angiotensinaldosterone system Increased secretion of glucagon and insulin Metabolism of catecholamines Urinary excretion Free noradrenaline (~0.5%) Conjugated with sulphate (~2%) Catechol O-methyl transferase (COMT) converts the catecholamines to metadrenaline and normetadrenaline forming about 3% of total excretion Monoamine oxidase (MAO) produces aldehydes that are immediately metabolised to the corresponding carboxylic acid or alcohol by aldehyde or alcohol dehydrogenases catalyses the metabolism of metadrenaline and normetadrenaline to vanilyl mandelic acid (VMA, ~65% of excretion) and the corresponding alcohol (methoxy hydroxy phenyl glycol (MOPG) ~35% of excretion) Dihydroxy phenyl glycol (DOPG) Noradrenaline released into the circulation is not converted to DOPG. Estimates of the excretion of non-metabolised catecholamines (i.e. adrenaline, noradrenaline , metadrenaline, normetadrenaline) form a better diagnostic test for pheochromocytomas. Measurement of the ratio of DOPG to noradrenaline concentrations in blood may be a more sensitive way of detecting pheochromocytomas Adrenal gland disorders Suki Sankaralingam Consultant Clinical Scientist MSc Essex University March 2010 Adrenal gland disorders Cushing’s syndrome Adrenal Insufficiency Congenital adrenal hyperplasia (CAH) Conn’s syndrome Phaeochromocytoma MSc Essex University March 2010 Cushing’s syndrome (CS) Most frequently seen in adults between the ages of 20 to 50 More common in women than men (5:1) Rarely, a patient may have an inherited gene mutation, such as Multiple Endocrine Neoplasia Type 1 or MEN-1, that increases risk of developing tumors throughout the endocrine system, including pituitary and adrenal tumors. Children with Cushing’s syndrome tend to be obese, develop slowly, and may remain short. Women may have excess hair on their face and chest and menstrual irregularities. Causes of Cushing’s syndrome Common (~ 99%) Uncommon (~ <1%) Exogenous therapeutic glucocorticoids Anterior pituitary adenoma Ectopic ACTH Adrenal adenoma Rare (~ <0.01%) Adrenal carcinoma Ectopic CRH Alcoholic Bilateral multinodular hyperplasia Clinical features of Cushing’s syndrome Signs and symptoms associated with Cushing’s syndrome vary but frequently include: Obesity in the torso with thinner arms and legs Large rounded face (moon face) Increased fat in the neck and shoulder area Thin fragile skin that bruises easily and heals slowly. Purplish streaks that look like stretch marks on their abdomen, thighs, and buttocks. Muscle weakness Osteoporosis High blood pressure Increased blood sugar MSc Essex University March 2010 Laboratory Investigations for CS Random cortisol and ACTH level – Not recommended. Should avoid identifying aetiology in the initial screening. 24 hour urine cortisol (or urine free cortisol) - overall cortisol production can be evaluated. At lease two measurements recommended due to variability in secretion. Relies on complete collection. Note: May be normal in cyclical on mild CS Falsely low- CrCl <60ml/min, false positive-over collection, excessive fluid intake Assay interference , reference range should be method specific. Late night salivary cortisol –Unbound biologically active, Less invasive, collection easy. Two measurements recommended. Not suitable for shift workers, variable sleep patterns False positive – licorise, tobacco use Not fully evaluated Dexamethasone Suppression Test - Dexamethasone is a synthetic steroid that mimics cortisol in the feedback inhibition of CRH and ACTH production. Patients with Cushing’s syndrome will not show adequate cortisol suppression after a single low dose of 1mg dexamethasone given between 23 and 24hrs and sample taken at 9.00am the following morning. Levels below 50nmol/L excludes CS False positive - oestrogen, OCP, anticonvulsants, rifampacin therapy, non compliance False negative – reduced clearance of dexamethasone in liver and renal failure Investigations for CS Higher doses of 2mg dexamethasone – 0.5mg, six hourly for 48 hours To distinguish between an true CS and other causes of Cushing’s syndrom (alcoholism, poorly controlled diabetes, depression) cont… Cortisol <50nmol/L in blood taken 6 hrs after last dose excludes CS Cortisol level at midnight – patient needs to be admitted and performed at least 48hrs after admission. CRH stimulation test – mostly used in specialised centres to locate difficult tumours Corticotrophin releasing hormone (CRH) is injected, and cortisol and ACTH levels are measured at baseline (before CRH) and at timed intervals after the injection, for example at 30 and 60 minutes. The normal response is a peak in ACTH levels followed by a peak in cortisol levels. Most patients with Cushing’s syndrome caused by adrenal tumors or ectopic ACTH-secreting tumors do not respond to CRH. ACTH levels may also be measured in samples obtained through a catheter placed in the inferior petrosal sinuses, which carry blood from the pituitary glands, and compared to blood ACTH concentrations. Computed tomography (CT) – used to help locate adrenal, pituitary, and ectopic tumours Magnetic resonance imaging (MRI) – sometimes ordered to help evaluate pituitary and adrenal glands Ultrasound Treatment for CS Treatment is to remove, block, or minimise the body’s exposure to excess cortisol. If due to corticosteroid use Minimize the dosage required. Never abruptly stop taking these medications - dosages must be changed slowly. Patients should consult with their family doctor or endocrinologist if required to adjust the dose to their needs. If due to a a single benign tumour or hyperplasia in one adrenal gland Surgical removal of the gland May need supplement because of atrophy of the remaining gland which will take some time to become fully functional. Nelson’s syndrome – caused by bilateral adrenalectomy. ACTH will be markedly elevated. If due to an ACTH-producing pituitary tumour Removal of the tumour will often resolve the excess cortisol. If removal not possible - radiation therapy If an ectopic ACTH-producing tumour(s) - surgery, radiation, and/or chemotherapy Addison’s disease Affects 1 to 4 people per 100,000 Found in patients of all ages Affects both males and females equally. Symptoms may not emerge until about 80% to 90% of the adrenal cortex has been destroyed. Causes of Addison’s disease Primary adrenal insufficiency (Addison’s) due to an autoimmune process (70%). other causes 30% Tuberculosis - common in areas where tuberculosis is more prevalent human immunodeficiency virus (HIV). Bacterial, viral (CMV) and fungal infections adrenal haemorrhage spread of cancer into the adrenal glands. rarely, it may be due to a genetic abnormality of the adrenal glands. Secondary adrenal insufficiency decrease production of the pituitary hormone ACTH due to pituitary damage, a pituitary tumour, or some other cause corticosteroid therapy (such as prednisone) is abruptly halted. With secondary adrenal insufficiency, aldosterone production is usually not affected. Clinical features of Adrenal Insufficiency The symptoms are non specific. They may emerge slowly, first appearing during times of stress, then increasing in intensity over a period of several months. Symptoms may include: Common Weakness (~100%) Weight loss (~100%) Pigmentation (~95%) Postural hypotension (~25%) Anorexia (~95%) Nausea (~95%) Abdominal pain (~30%) Uncommon Vitiligo (~20%) Salt craving (~15%) Hypoglycemia (in adults ~ <1%) Aches and pains (~10%) About 25% of the time, adrenal insufficiency is diagnosed during an adrenal crisis (also called an Addisonian crisis). This crisis may be caused by a period of increased stress, trauma, surgery, or a severe infection. If left untreated it can be fatal. Signs and symptoms may include: Kidney failure Loss of consciousness Low blood pressure Severe pain in the lower back, abdomen or legs Severe vomiting and diarrhea, leading to dehydration Shock Laboratory Investigations To determine whether adrenal insufficiency is present To distinguish between primary and secondary insufficiency To determine the underlying cause Electrolytes – Low sodium (70%) High potassium (35%) Glucose level - Low Renal function – Pre uraemic Cortisol at 9.00am low levels confirms the suspicion normal level does not exclude very high levels excludes adrenal insufficiency. If the adrenal gland is either not functioning normally or not being stimulated by ACTH, then cortisol levels will be consistently low. Cortisol levels are used, along with ACTH and ACTH stimulation tests, to help diagnose adrenal insufficiency. Adrenal antibodies - good marker of autoimmune Addison's disease. Not routinely done Renin activity - elevated in primary adrenal insufficiency lack of aldosterone causes increased renal sodium losses. This lowers blood sodium levels and decreases the amount of fluid in the blood (which lowers blood volume and pressure), which in turn stimulates renin production by the kidney. Laboratory Investigations –cont… Synacthen stimulation test Measuring levels of cortisol in blood before and 30 and 60 minutes after an injection of 250µg im synthetic ACTH If the adrenal glands are functional - cortisol levels will rise in response to the ACTH stimulation (200nmol above the basal concentration or the final concentration of at lease 550nmol) If they are damaged or non-functional - response to ACTH will be minimal. ACTH - baseline test to evaluate whether or not the pituitary is producing appropriate amounts of ACTH. low ACTH levels indicate secondary adrenal insufficiency high levels indicate primary adrenal insufficiency (Addison’s disease). Other investigations and treatment X-rays Calcification on the adrenal cortex -may be due to a tuberculosis CT or MRI Enlarged adrenal glands -infections, cancers Normal or small size adrenal glands -autoimmune disease , secondary adrenal insufficiency If the condition is due to an infection may regain some adrenal function when the infection resolves replacing the missing hormones (hydrocortisone, fludrocortisone) In the case of secondary adrenal insufficiency - hormone replacement. Once suspected, it is imperative that Addison's disease is confirmed biochemically and treated immediately. Congenital adrenal hyperplasia 21-hydroxylase (CYP21A2) deficiency - most common ( 95%) Between 1 in 5000 and 1 in 15 000 live births in western countries Clinical manifestations - loss of aldosterone and cortisol metabolism with precursors being shunted into androgen synthesis (vary according to the sex of the patient) Total ablation of enzyme activity (e.g. deletion or nonsense mutations) results in ‘saltwasting’ disease (loss of aldosterone) and virilization and ambigous genitalia of a female infant (increased testosterone production). Salt wasting results in severe dehydration in the first 14 days of life with hypotension and death if untreated Mutations resulting in 1–2% normal enzyme activity (e.g. missense mutations) have virilization but not salt-wasting Mutations resulting in 20–60% normal enzyme activity give the so-called ‘non-classical’ presentations Boys without salt-wasting may present with precocious sexual development Treatment Glucocorticoid therapy (monitored to result in suppression of the high concentrations of 17αhydroxyprogesterone) Mineralocorticoids (monitored by blood pressure and by assays of plasma renin) Congenital adrenal hyperplasia –cont… 11 beta-hydroxylase (CYP11B1) deficiency 17 alpha-hydroxylase/17,20 lyase (CYP17) deficiency Extremely rare (approximately 200 cases in the world literature) and expressed in both adrenal gland and gonad. Clinical presentation: hypertension with hypokalemia due to excessive production of mineralocorticoids; failure of pubertal development in genetic females and genetic males presenting at puberty with female external genitalia and intra-abdominal testes 3 beta-hydroxysteroid dehydrogenase (3β-HSD2) deficiency Very rare incidence of ~1 in 100 000 live births Clinical presentation: virilisation, similar to CYP21A2 deficiency but with additional hypertension, perhaps due to increased production of 11-deoxycortisol which has mineralocorticoid actions Classical form leads to defective production of all steroids. Clinical presentation: adrenal failure in early infancy; moderate virilization in females; varying degress of genital ambiguity in males; mild, non-classical form may present with hirsutism and oligomenorrhea Side-chain cleavage enzyme; desmolase (CYP11A1) and StAR protein defect: Loss of all steroidogenic capacity in adrenal gland and gonad. Clinical presentation: adrenal failure in early infancy; genetic males have female external genitalia (loss of androgens); frequently fatal if undiagnosed CAH OxidoReductase Deficiency (ORD) –another variant of CAH (first reported in 2004) Mutation in the electron donor enzyme P450 oxidoreductase (POR) cont….. POR is the electron donor for all microsomal P450 enzymes, including three steroidogenic enzymes 450c17(17alpha hydroxylase/17,20-lyase), P450c2(21-hydroxylase) and P450aro(aromatase). Partial deficiency of 21hydroxylase and 17alpha hydroxylase Cortisol deficiency – clinically insignificant to life threatening Two unique features (not seen in other CAH variants)-skeletal malformation and severe genital ambiguity in both sexes Clinical manifestation of ORD Clinical diagnosis Ambiguous genitalia in both males and females Primary amenorrhoea and cystic ovaries in females Poor masculinisation during puberty in males Maternal virilisation during pregnancy with an affected fetus. Apparently healthy infant with 21-hydroxylase deficiency on newborn screening Whose mothers were virilised during pregnancy Biochemical diagnosis Detection of steroid abnormalities using GC-MS Clinical spectrum of 21-OH deficiency Newborn Ambiguous genitalia in a female Absent testes in an apparent male Severe shock Failure to thrive Isolated clitoromegaly Isolated labial fusion Childhood Premature adrenarche Tall stature and advanced bone age Adult Menstrual irregularities Hirsutism Short stature Obesity Male infertility Testicular tumours Conn’s syndrome Most common cause of secondary hypertension Characterized by excessive secretion of aldosterone from the adrenal glands. Also referred to as primary hyperaldosteronism Excessive aldosterone is produced by One or more benign adrenal tumours Hyperplasia Glucocorticoid – suppressible hyperaldosteronism (autosomal dominant- ACTH dependent) Idiopathic cancerous adrenal tumour (rare) Commonly occurs in adults between the ages of 30 and 50 (although it can be in anyone) More common in women than men Presence of hypokalemia with hypertension - suggests possible primary hyperaldosteronism. Resistant to standard hypertension therapies – suspicion of primary hyperaldosteronism is high Clinical and Laboratory findings Hypertension (non responsive to 2-3 antihypertensive drugs) Hypokalemia and inappropriate kaliuria Alkalosis Hypernatraemia (rare) Nonspecific symptoms frequent urination, increased thirst, weakness, fatigue, temporary paralysis, palpitations, headaches, muscle cramps, and tingling. Investigations for Conn’s Diagnosing Conn’s syndrome is important Few causes of hypertension that is potentially curable Secondary aldosteronism must be distinguished from primary aldosteronism. The ratio of aldosterone to renin is used to screen for primary hyperaldosteronism. Patients should be on adequate sodium (100-150 mmol/day) and potassium (50 – 100 mmol/day) prior to test Potassium supplement should be stopped 24hrs prior to the test( spironolactone must be stopped for 6weeks} should be off ACE inhibitors, Beta blockers and calcium channel blockers and diuretics,. Sample should be sent to lab within 30 minutes of venepuncture at RT Separated and frozen immediately and kept frozen until analysis for renin) Low renin and high aldosterone - significantly increased ratio (>2000) Consistent with primary hyperaldosteronism Ratio <800 –unlikely of Conn’s Investigations for Conn’s – cont…. CT/MRI scan of the adrenal glands benign adrenal tumours are relatively common Many of them do not secrete aldosterone and are found incidentally during procedures for other reasons. Determining hyperplasia can also be tricky because the size of normal adrenal glands may vary significantly from one person to the next. If negative CT – saline suppression test to locate the tumour when positive lab result and high clinical suspicion. If hyperplasia or an aldosterone-producing tumour is suspected adrenal venous sampling Tested for aldosterone, cortisol and aldosterone/ cortisol ratio calculated. Results from the two adrenal glands compared. If significantly different - adenoma located in the gland with the highest aldosterone concentration. Treatment for Conn’s Lower blood pressure to normal or near normal levels Decrease blood levels and resolve electrolyte imbalances If due to a single benign adrenal tumour – surgical removal. If the primary hyperaldosteronism is due to a cancerous tumour (rare) If hypertension does not resolve additional therapy to control BP organs located next to the affected adrenal gland will need to be evaluated during surgery and more than the adrenal gland may need to be removed. If the cause is idiopathic or appears to be due to hyperplasia in both adrenals surgery not recommended Treatment with drugs to block the action of aldosterone and with one or more blood pressure drug therapies. Mineralocorticoid deficiency Biochemical features Hyponatraemia Hyperkalaemia Hypercholoraemic metabolic acidosis Hypovolaemia Causes Aldosterone synthase deficiency () Combined with glucocorticoid deficiency CAH-21 hydroxylase, 3HSD and desmolase deficiency (autosomal recessive) Adrenoleukodystrophy – X linked Autoimmune TB, AIDS Phaeochromocytoma Rare tumours (1 per million per annum) Usually benign Found in the sexes equally Incidence between the ages of 20 and 50 years although can occur at any age In general, 10% are bilateral, 10% are extra-adrenal, 10% occur in childhood 10% are malignant. The majority of pheochromocytomas are sporadic and without known cause. Some occur in MEN type 1 Clinical features of phaeochromocytoma Common Headache (~60%) Palpitations (~60%) Anxiety (~50%) Sweating (~50%) Abdominal pain (~25%) Glucose intolerance or diabetes mellitus (~40%) Hypertension - sustained or paroxysmal with or without postural hypotension (~50%) Uncommon Weight loss (~10%) Chest pain (~20%) Tremor (~5%) Pallor (~5%) Investigations Analysis of catecholamine by HPLC with electrochemical detection Urinary and plasma catecholamine metabolites, metanephrines and normetanephrines - lowest false negatives. Several stimulation and suppression tests are also available but the safest are the glucagon stimulation and the clonidine or pentolinium suppression tests. Catecholamine secretion from a pheochromocytoma (but not normal adrenal medulla) is stimulated approximately 2–5-fold by glucagon catecholamine secretion from a pheochromocytoma is not suppressed by clonidine or pentolinium. These drugs suppress catecholamine secretion by at least 50% from a normal adrenal medulla. MIBG(meta iodo benzyl guanine) – To locate the tumour Case studies in adrenal gland disorders Suki Sankaralingam Consultant Clinical Scientist MSc Essex University March 2010 Case 1 A young female presented to her GP with hirsutism, amenorrhoea for 4 months and weight gain Testosterone = 7.9 nnmol/L (0.9 – 3.6) DHEAS = 14.7 µmol/L (1.2 – 11.0) UFC= 1740 nmol/L Dexamethasone suppression = cortisol 900nmol/L CT of the abdomen : mass in the left adrenal. Proved to be carcinoma of the adrenal cortex Case 2 65yr old man was seen in A/E with weight loss, pigmentation and respiratory distress Na 144mmol/L K 2.0 mmol/L HCO3 >40 mmol/L Urea 8.6 mmol/L Creat 120 µmol/L Cortisol >1600 mmol/L ACTH 550 ng/L (5-60) Diagnosis: ectopic ACTH , oat cell carcinoma of the lungs MSc Essex University March 2010 Case 3 A 48yr old truck driver, who has never been to a GP, presented to the casualty complaining of abdominal pain, extreme tiredness and recent history of vomiting. Na 119 K 5.4 Urea 16.0 Creat 182 Glucose 4.0 Short synacthen 260nmol/L(basal); 282 (30mins); 285(60 mins) Diagnosis: Addison’s Adrenal antibodies : Positive Autoimmune Thyroid antibodies : Positive , Thyroid function : Subclinical hypothyroidism MSc Essex University March 2010 Case 4 40 year old female presented with blood pressure of 178/120 mmHg. She has been on antihypertensive medication including diuretics for 6 months, but her BP remained uncontrollable. Blood results 2 weeks after stopping the medication Na 145 mmol/L (132 -144) K 2.6 mmol/L (3.2 -4.8) Cl 95 mmol/L (98 -108) HCO3 35 mmol/L (23-33) Urine K = 75mmol/L hypokalaemic metabolic alkalosis and inappropriate urinary potassium loss ?Primary hyperaldosteronism Random Plasma Renin <0.01 pmol/ml/hr (2.1-4.7); Aldosterone 900pmol/L (110-860) ARR >1000 Adrenal Tumour on CT MSc Essex University March 2010 Case 5 12yr male child was admitted with epileptic fits and vomiting over the past 12 hrs. He was normal prior to that. On examination there was no indication of meningitis. His BP 180/120 mm Hg and pulse rate 92 min. Blood Na = 135mmol/L; K= 4.1 mmol/L; Cl = 98 mmol/L; HCO3 = 25 mmol/L; mmol/L; Creat =60µmol/L; Glucose=10.5 mmol/L Urine Na = 50mmol/L; Potassium =40 mmol/L CSF Total Protein = 0.43 g/L (0.1 – 0.4); Glucose = 6.2 mmol/L (1.7 -3.9) BP varied between 160/120 and 125/80 Differential diagnosis: Meningitis, encephalitis, hypertension. Renal angiogram revealed: stenosis of the left renal artery PRA = 10.2 pmol/ml/hr (2.1–4.7) and Aldosterone = 980pmol/L (110-860) Consistent with secondary hyperaldosteronism Before the surgical correction, a mass on the left kidney was noticed by the surgeon. Urine metadrenaline = 5.0µmol/24hrs (>5.0 for adults) Further estimation 9.8 and 8.3 Indicated presence of Phaeochromocytoma which was surgically removed. MSc Essex University March 2010 Urea =4.8 Case 6 Two weeks old male infant admitted to hospital severely dehydrated and critically ill Na = 106mmol/L K = 6.6mmol/L Cl = 70mmol/L HCO3 = 19mmol/L Urea = 6.5mmol/L Creat = 50µmol/L 17-hydroxy progesterone = 950 nmol/L Very high 17OHP - ? 21 hydroxylase or 11 beta hydroxylase deficiency Hyperkalaemia and hyponatraemia – consistent with 21 hydroxylase deficiency. MSc Essex University March 2010 Case 7 44 year old short male was incidentally discovered with bilateral adrenal tumours on CT examination for flank pain. The tumours were non malignant. On subsequent presentation his blood results showed PRA of 12.8 pmo/ml/hr (2.1-4.7); Aldosterone 1280 pmol/L (110-860) BP 134/80 mmHg, Skin was pigmented, no abnormalities on penis size or testicular volume Electrolytes were within the reference range ACTH : 149 ng/L (5-60) ; Cortisol 314 nmol/L Urinary steroid profile : Elevated excretion of ketosteroids except 17hydroxy steroids Dexamethasone suppression and ACTH stimulation tests were performed. Progesterone and 17OHP were markedly elevated. DHEAS and androstenedione were elevated. 11 deoxycortisol and cortisol were within reference while 11 deoxycorticosterone and aldosterone were elevated Results confirmed mild form of CAH Short stature – due to precocious puperty Pigmentation – hypersecretion of ACTH The tumours were due to ACTH ACTH suppression therapy prevented further growth and to normalise PRA MSc Essex University March 2010 Case 8 33yr old married man was prescribe hydrocortisone (HC) for ulcerative colitis He started developing gynaecomastia and was referred for endocrine opinion Gonadotrophins, Oestrogen and androgens were measured. High oestrogen; FSH/LH were suppressed; Tetosterone was also high He started menstruating 17OHP was very high with high androgens Confirmed CAH – 21 hydroxylase deficiency When he was on HC, it has suppressed the ACTH which as a result reduced the adrenal androgens. The ovary then started producing oestrogen. MSc Essex University March 2010 Case 9 26yr old waitress was referred to the endocrine clinic for excessive hair growth. Has been a problem since the age of 16yrs and has got worse and shaving every other day. Irregular periods and could be without it for 5 months On examination: BP 120/78; BMI 28.4; prominent facial hair and acne; no virilism TSH=2.1mu/L; FT4 =16.6 pmol/L; Prolactin=232mU/L; FSH=7.4U/L; LH=25.4U/L; Testosterone = 3.4nmol/L; SHBG=35nmol/L; Oestradiol = 256pmol/L; UFC=322nmol/24hr What is the diagnosis? PCO or late onset CAH What test would you do to confirm/rule out LO-CAH? Synacthen Stimulation test and measure 17OHP on all three samples Basal 17OHP = 6nmol/L 30mins post synacthen = 35nmol/L 60mins post synacthen = 38nmol/L Confirms late onset CAH