Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Designer baby wikipedia , lookup
Gene expression profiling wikipedia , lookup
Pathogenomics wikipedia , lookup
Microevolution wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Non-coding DNA wikipedia , lookup
Human genome wikipedia , lookup
Microsatellite wikipedia , lookup
Point mutation wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Genome editing wikipedia , lookup
Metagenomics wikipedia , lookup
Smith–Waterman algorithm wikipedia , lookup
Computational phylogenetics wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Helitron (biology) wikipedia , lookup
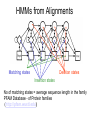
Profiles for Sequences Sequence Profiles • Often, sequences are characterized by similarities that are not well captured through matching algorithms. • For example, identification of genes in the presence of exons/introns, gene features (CpG islands, etc.), domain profiles in proteins, among others. • For such sequences, Markov chains provide useful abstractions. Markov Chains Rain Sunny Cloudy State transition matrix : The probability of States : Three states - sunny, the weather given the previous day's cloudy, rainy. weather. Initial Distribution : Defining the probability of the system being in each of the states at time 0. Hidden Markov Models Hidden states : the (TRUE) states of a system that may be described by a Markov process (e.g., the weather). Observable states : the states of the process that are `visible’. 4 Hidden Markov Models Initial Distribution : Initial state probability vector. State transition Matrix Emission Probabilities: containing the probability of observing a particular observable state given that the hidden model is in a particular hidden state. Hidden Markov Models Transition Prob. Output Prob. Observed sequences can be scored if their state transitions are known. The probability of ACCY along this path is: .4 * .3 * .46 * .6 * .97 * .5 * .015 * .73 *.01 * 1 = 1.76x10-6. Methods for Hidden Markov Models Scoring problem: Given an existing HMM and observed sequence , what is the probability that the HMM can generate the sequence Methods, contd. Alignment Problem Given a sequence, what is the optimal state sequence that the HMM would use to generate it Methods, contd. Training Problem How do we estimate the structure and parameters of a HMM from data. HMMs– Some Applications • • • • • Gene finding and prediction Protein-Profile Analysis Secondary Structure prediction Copy Number Variation Characterizing SNPs Gene Template (Left) (Removed) 11 HMMs: Applications • Classification: Classifying observations within a sequence • Order: A DNA sequence is a set of ordered observations • Structure : can be intuitively defined: • Measure of success: # of complete exons correctly labeled • Training data: Available from various genome annotation projects HMMs for Gene Finding • Training - Expectation Maximization (EM) • Parsing – Viterbi algorithm An HMM for unspliced genes. x : non-coding DNA c : coding state Genefinders: a Comparison Accuracy per nucleotide Method Sn Sp AC Sn GENSCAN FGENEH GeneID GeneParser2 GenLang GRAILII SORFIND Xpound 0.93 0.77 0.63 0.66 0.72 0.72 0.71 0.61 0.93 0.85 0.81 0.79 0.75 0.84 0.85 0.82 0.91 0.78 0.67 0.66 0.69 0.75 0.73 0.68 0.78 0.61 0.44 0.35 0.5 0.36 0.42 0.15 Accuracy per exon (Sn+Sp)/ Sp ME 2 0.81 0.8 0.09 0.61 0.61 0.15 0.45 0.45 0.28 0.39 0.37 0.29 0.49 0.5 0.21 0.41 0.38 0.25 0.47 0.45 0.24 0.17 0.16 0.32 Sn = Sensitivity Sp = Specificity Ac = Approximate Correlation ME = Missing Exons WE = Wrong Exons GENSCAN Performance Data, http://genes.mit.edu/Accuracy.html WE 0.05 0.11 0.24 0.17 0.21 0.1 0.14 0.13 Protein Profile HMMs • Motivation – Given a single amino acid target sequence of unknown structure, we want to infer the structure of the resulting protein. Use Profile Similarity • What is a Profile? – – – – Proteins families of related sequences and structures Same function Clear evolutionary relationship Patterns of conservation, some positions are more conserved than the others HMMs From Alignment ACA TCA ACA AGA ACC - - - ATG ACT ATC C - - AGC - - - ATC G - - ATC insertion Transition probabilities Output Probabilities A HMM model for a DNA motif alignments, The transitions are shown with arrows whose thickness indicate their probability. In each state, the histogram shows the probabilities of the four bases. HMMs from Alignments Deletion states Matching states Insertion states No of matching states = average sequence length in the family PFAM Database - of Protein families (http://pfam.wustl.edu) Database Searching • Given HMM, M, for a sequence family, find all members of the family in data base. • LL – score LL(x) = log P(x|M) (LL score is length dependent – must normalize or use Z-score) 18 Querying a Sequence Suppose I have a query protein sequence, and I am interested in which family it belongs to? There can be many paths leading to the generation of this sequence. Need to find all these paths and sum the probabilities. Consensus sequence:ACAC - - ATC P (ACACATC) = 0.8x1 x 0.8x1 x 0.8x0.6 x 0.4x0.6 x 1x1 x 0.8x1 x 0.8 = 4.7 x 10 -2 Multiple Alignments • Try every possible path through the model that would produce the target sequences – Keep the best one and its probability. – Output : Sequence of match, insert and delete states • Viterbi alg. Dynamic Programming 20 HMMs from Unaligned Sequences • Baum-Welch Expectation-maximization method – Start with a model whose length matches the average length of the sequences and with random output and transition probabilities. – Align all the sequences to the model. – Use the alignment to alter the output and transition probabilities – Repeat. Continue until the model stops changing • By-product: a multiple alignment PHMM Example An alignment of 30 short amino acid sequences chopped out of a alignment of the SH3 domain. The shaded area are the most conserved and were represented by the main states in the HMM. The unshaded area was represented by an insert state.