Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Bisulfite sequencing wikipedia , lookup
Gene regulatory network wikipedia , lookup
Gene desert wikipedia , lookup
Expression vector wikipedia , lookup
Promoter (genetics) wikipedia , lookup
Transcriptional regulation wikipedia , lookup
Copy-number variation wikipedia , lookup
Gene expression wikipedia , lookup
Point mutation wikipedia , lookup
Exome sequencing wikipedia , lookup
Zinc finger nuclease wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Genetic engineering wikipedia , lookup
Two-hybrid screening wikipedia , lookup
Transposable element wikipedia , lookup
Silencer (genetics) wikipedia , lookup
Homology modeling wikipedia , lookup
Personalized medicine wikipedia , lookup
Community fingerprinting wikipedia , lookup
Non-coding DNA wikipedia , lookup
Endogenous retrovirus wikipedia , lookup
Whole genome sequencing wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Genomic library wikipedia , lookup
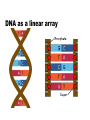
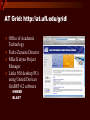
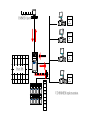
Using the AT Grid for Genomics Research at the University of Florida big biology meets obvious opportunity Interdisciplinary Center for Biotechnology Research Established at the University of Florida in 1987 by the Florida Legislature centralized organization of biomedical core facilities supporting biotechnology-based research Organized under the office of the Vice President of Research Genomics group founded in 1998 to begin providing large-scale DNA sequencing services ICBR Genomics Group Bill Farmerie Mick Popp, David Moraga, Sharon Norton, Li Zhang Gene Expression Core Li Liu, Fahong Yu, Brian Dill Scientific Director ICBR Genomics Group Bioinformatics Core Ernie Almira, Regina Shaw, Neda Panayotova, Kevin Holland, Patrick Thimote, Tina Langaee, Stephen Marsh Genomics Core What we do Large-scale DNA sequencing projects Microarray gene expression analysis Bioinformatics Resource Faculty research programs On campus Satellite research facilities Other SUS Universities Biotech industry Exploring Genome Space Biology moves from a data-poor to a data-rich science The Genome Covers The Human Genome Project HGP drives innovation 2 major technological benefits stimulated development of high throughput methods computational tools for data mining and visualization of biological information Genbank August 15 2004 37,343,937 loci 41,808,045,653 bases 37,343,937 reported sequences Growth of DNA Sequence Output What is this curious relationship between genes and computation? It is all about information management Bioinformatics the intersection of biology and information sciences Informational Macromolecules Living things store, search, and selectively retrieve biological information DNA the structure A:T G:C DNA as a linear array we store lots of information using simple linear codes CTGGGTTCTGTTCGGGATCCCAGTCACAGGGACAATGGCGCATTCATATGTCACTTCCTTTACCTGCCTGGAG GAGGTGTGGCCACAGACTCTGGTGGCTGCGAACGGGGACTCTGACCCAGTCGACTTTATCGCCTTGACGAAGG GTTGGTTAATCCGTGCATGTGAGCTCCTCAGGGTGGAATCCAGGAGGATCCACGAGGGTGAATTGGCGGCATT CTTGTCTTACGCCATCGCCTACCCCCAAAACTTCCTGTCTGTGATTGACAGCTACAGCGTAGGATGCGGTCTG TTGAACTTCTGCGCGGTGGCTCTGGCTCTCTGTGAACTGGGCTACAGGCCTGTGGGGGTGCGTTTGGACAGCG GTGACCTCTGCAGCCTGTCGGTGGATGTCCGCCAGGTCTTCAGACGCTGCAGCGAGCATTTCTCCGTCCCTGC CTTTGATTCGTTGATCATCGTCGGGACGAATAACATCTCAGAGAAAAGCTTGACGGAGCTCAGCCTGAAGGAG AACCAGATTGACGTTGTCGGAGTCGGAACTCACCTGGTCACCTGTACGACTCAGCCGTCGCTGGGTTGCGTTT ACAAGCTGGTGGAGGTGAGGGGGAGGCCCCGGATGAAGATCAGCGAGGATCCGGAAAAGAGCACCGTTCCCGG GAGGAAGCAGGTGTACCGCCTGATGGACACTGATGCTCCTCCAGAACCTGGAGTCCCTCTGAGCTGCTTCCCT CTGTGCTCCGATCGCTCCTCCGTCTCCGTCACCCCGGCGCAGGTTCACCGTCTGCGGCAGGAAGTCTTTGTTG ATGGACAGGTCACAGCCCGTCTGTGCAGCGCCACAGAGACCAGAACGGAGGTCCAGACCGCTCTCAAGACCCT CCACCCTCGACACCAGAGGCTGCAGGAGCCAGACTCGTACACGGTGATTCACATTCTGAAGAAAACAACATTG GATCGCGCTTTTCCGCTCTCTTCCCTTAGTTTCCCCTCCGAACTCCGCCGCTGGGCCGGAGGACTGAACCGGC CCCCGACGGTGTCCCAGCGGCGGTGCAATGTGGCCCGGGTCCGGGAGGAGTGCGTGACGCCAGAGCAGAATGG TTCGGTGGACGGGGGCGCACACGCTTCTCGCCGCGGCCGCTCCCCGCGGCCCACGGAACCGCGGGATCGGAGC TGTTTTGTGCCGCCTGAAGGACTCGAAGGGGGACGGATAAATGCTGGATCCCCGAGTCCAGATCTGACCGTCT GCATTCCGCTGGTGAGCTGCCAGACGCATCTGGAAACGAGCGCCGACAGAAGCAGCTCCGGACCATGTCGCCG TCCGCGCACACAGGTCGCGTGTAAAGGGGACTTGGTCAGATCATCTTGCACCGGAACCAGGTCTCCCCTGGAG ATGGGGACGGTCATGACCGTCTTCTACCAGAAGAAGTCCCAGCGGCCGGAGAGGAGAACCTTCCAGATCAAGC CTGACACGCGGCTCCTCGTGTGGAGCCGAAACCCCGACAAAAGCGAAGGAGAGAGTGAGTATGAGCAGGCGGG CCGTGCCGGGACCGGGCCCACGCCGCCCAGAACCTCATGTTCCTGGTGTTCCAGCACCGACCGGCCAGTTCTG GCTCAGCTCCACACAACATCTGACAAACCCTCGTGGTTCCTGGTGGTCGACCACACGGCTGGTGAGGCGGCCT CAGGTAGCTCAGGTAGCTCAGGTTAGCGTAAAGGGAGTTTTAAGCATCACCTGGTGACGGGGCAGGTGAGCTC CAGCCACTCAGCAGTGCACGGCCGTGCACATACACACACACCTCTGTGTCGAGGTTACAGGTGGGGCCAAAGC CCAACACCTTCAATGGCCCTCAGAGCTTTGAGGTTTTGAGGAATTGAGCCTTTAATCAGAAAA another simple linear code is the basis of life Biological Information information storage selective information retrieval function From Sequence to Function The genomic sequence identifies the 'parts' the next trick is understanding gene function Post genomic era = functional genomics Critical concept: genes of similar sequence may have similar functions Inferring function for a new gene begins with searching for it’s nearest neighbor (or homolog) of known function BLAST Most common starting point for gene identification Input: nucleotide or amino acid sequence Similarity search of sequence repository (GenBank) Output Calculated scores (bit score and e-value) Text string (definition line), ID Reference Tag Sequence alignment Advantages Fast algorithm, very good at finding close homologs Disadvantages Only finds genes existing in the search database Not good at finding distant relatives Alternatives to BLAST HMMER developed by Sean Eddy Uses Hidden Markov Models Searches unknown protein query sequence against a database of protein family models Statistical models constructed from alignment of conserved protein regions (Pfam) 7677 families in Pfam release 16.0 Advantages Superior to BLAST for discovering more distant homology relations Disadvantages More computationally intensive than BLAST This could be a super computer AT Grid: http://at.ufl.edu/grid/ Office of Academic Technology Fedro Zazueta Director Mike Kutyna Project Manager Links 500 desktop PCs using United Devices GridMP 4.2 software HMMER BLAST Blast Query HMMER Query Query 1 Genome DB segment Query 1 Genome DB segment Query 1 Results Genome DB segment Query 1 Genome Database Pfam DB Genome segments Query 1 Query 1 Query 1 Genome DB segment Genome Query 1 segment Genome segment Genome segment Genome segment Genome UD NCBI Blast Implementation UD HMMER implementation Sample HMMER Output Query: 7.UF_CU.3.CB366 804 1 630 nseq=40; translated Scores for sequence family classification (score includes all domains): Model Description Score E-value N -------- --------------- ------- --KH_1 KH domain 127.6 2.3e-34 2 KH_2 KH domain 3.0 0.88 2 Parsed for domains: Model Domain seq-f seq-t hmm-f hmm-t score E-value -------- ------- ----- ----- ----- --------- ------KH_2 1/2 36 93 .. 1 78 [] 3.4 0.8 KH_1 1/2 36 98 .. 1 74 [] 66.9 4.4e-16 KH_2 2/2 118 160 .. 1 78 [] 0.1 1.8 KH_1 2/2 118 180 .. 1 74 [] 68.3 1.6e-16 Alignments of top-scoring domains: KH_2: domain 1 of 2, from 36 to 93: score 3.4, E = 0.8 *->avivvirtsrpGivIGKgGsnIkklgkelrklltgkkvqieviEySd i + ++ G +IG+gGs I+ l+++ + ++i E 7.UF_CU.3. 36 SDIMMVESANVGKIIGRGGSKIRDLEQDSNAR-----IKISRDE--- 74 eeFgkkVfLeLwVKVkknWvknpellaqLga<-* +++ v+ ++ ++ a 7.UF_CU.3. 75 ---DENGMKS---------VEISGTDEEIDA 93 KH_1: domain 1 of 2, from 36 to 98: score 66.9, E = 4.4e-16 *->terilippskvgriIGkgGstIkeIreetGakIdipddgseskplpe + + + + vg+iIG+gGs+I+ ++++++a+I+i++d+ 7.UF_CU.3. 36 SDIMMVESANVGKIIGRGGSKIRDLEQDSNARIKISRDE-------- 74 dplngsdertvtIsGtpeavekAkkli<-* + +++ + v+IsGt e++++Ak++i 7.UF_CU.3. 75 -D--ENGMKSVEISGTDEEIDAAKRMI 98 Good news -- Bad news AT Grid compresses time for HMMER searches ~100X Accepts batch queries as input Query sequences must be pre-computed protein translation Requires additional step & CDS prediction Output is flat text Developed Perl-based parser Tabbed output as input to relational DB Integrating gene annotation data from as many applications as possible Facilitates comparison of results for the same query