Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Drug interaction wikipedia , lookup
NMDA receptor wikipedia , lookup
Discovery and development of beta-blockers wikipedia , lookup
Discovery and development of proton pump inhibitors wikipedia , lookup
Toxicodynamics wikipedia , lookup
Polysubstance dependence wikipedia , lookup
Discovery and development of cephalosporins wikipedia , lookup
5-HT2C receptor agonist wikipedia , lookup
5-HT3 antagonist wikipedia , lookup
Discovery and development of TRPV1 antagonists wikipedia , lookup
CCR5 receptor antagonist wikipedia , lookup
Dextropropoxyphene wikipedia , lookup
Discovery and development of ACE inhibitors wikipedia , lookup
Psychopharmacology wikipedia , lookup
Drug discovery wikipedia , lookup
Discovery and development of angiotensin receptor blockers wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Neuropharmacology wikipedia , lookup
Discovery and development of antiandrogens wikipedia , lookup
Nicotinic agonist wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
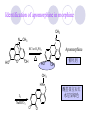
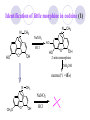
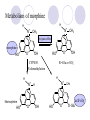
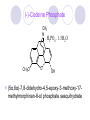
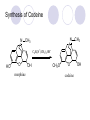
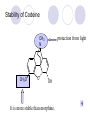
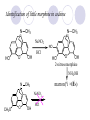
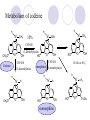
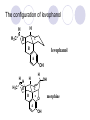
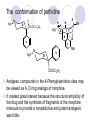
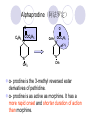
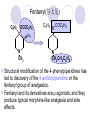
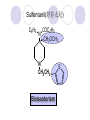
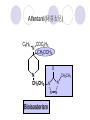
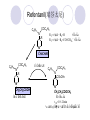
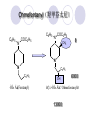
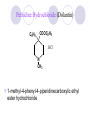
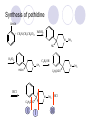
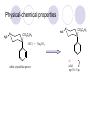
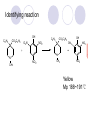
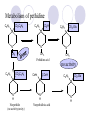
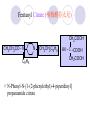
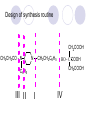
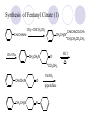
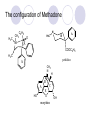
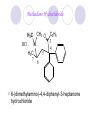
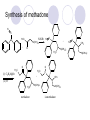
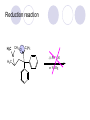
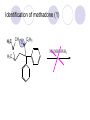
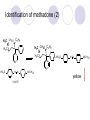
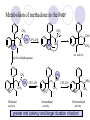
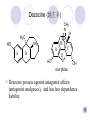
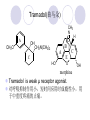
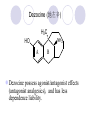
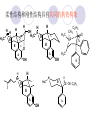
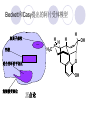
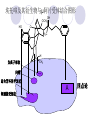
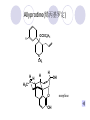
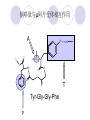
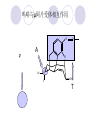
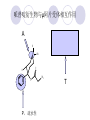
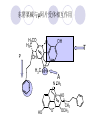
Chapter 7 Analgesics Analgesics Agents that decrease pain are referred to as analgesics or as analgetics(止痛药). Drugs used to relieve pain include: the nonsteroidal anti-inflammatory agents anesthetics (general or local) central nervous system (CNS) depressants opioid agents Opioid agents Opioid analgesics historically have been called narcotic (麻醉性) analgesics. Narcotic analgesic literally means that the agents cause sleep or loss of consciousness in the conjunction with their analgesic effect. The term narcotic has become associated with the addictive properties of opioids and other CNS depressants. Because the great therapeutic value of the opioids is their ability to cause analgesic without causing sleep (narcosis昏迷状态),the term narcotic analgesic is not used further in this chapter. Opium alkaloids The use of the juice (opium in Greek) or gum from the unripe seed pods of the poppy (罂粟) is among the oldest recorded medications. The pharmacist Surturner first isolated an alkaloid from opium in 1803. He named the alkaloid morphine, after the Greek god of dreams. Codeine, thebaine(蒂巴因), and papaverine (罂粟碱) are other medically important alkaloids that were later isolated from opium gum. Morphine and its analogous(类似物) Morphine was among the first compounds to undergo structure modification. Ethylmorphine (the 3-ethyl ether of morphine ) was introduced as a medicine in 1898. Diacetylmorphine, which may be considered to be the first synthetic prodrug, was synthesized in 1874 and introduced as a nonaddicting analgesics, antidiarrheal(止泻剂), and antitussive agent (止咳药) in 1898. Opiate The term opiate was used extensively until the 1980s to describe any natural or synthetic agent that was derived from morphine. An opiate was any compound that was structurally related to morphine. Opioid The discovery, in the mid-1970s, of peptides in the brain that had pharmacologic actions similar to those of morphine prompted a change in nomenclature (命名). The peptides were not easily related to morphine structurally; yet, their actions were similar to the actions of morphine. The term opioid, meaning opium-like or morphine-like in terms of pharmacologic action, was introduced. Opioids include: alkaloid opioids,such as morphine; synthetic analgesics, such as pethidine; endogenous opioids, such as enkephalins opioid antagonists, such as naloxone. Principal pharmacologic effects of opioids (-)-Morphine and its congeners modify the effects of pain impulses on the CNS. Awareness of the pain may persist or diminish but the ability to interpret, integrate, and react to pain is decreased with attendant sedation, euphoria(欣 快), and reduced anxiety and suffering. The only other useful effect of opioids on the CNS is cough suppression. Side effect of Opioids Constipation(便秘) Respiratory depression, which is the cause of death from opioid overdose Dependence Tolerance, which refers to the need to increase the dose of opioid over a period of time in order to achieve the same level of analgesia or euphoria. 吗啡连续反复使用,易产生耐受性和成瘾性,须 严格按照国家颁布的《麻醉药品管理办法》管理。 Opioid receptors Narcotic analgesics(agonists and antagonists) are stereospecific binding, chemically specific binding, and competitive binding with opioid receptors. Opioid receptors are classified: μ:μ1、μ2 κ:κ1、κ2、κ3 δ:δ1、δ2 Section 1 Morphine and related opioid agonists (1)Opioid alkaloid (2)Morphine’s derivatives (3)Synthetic analgesics (1) opioid alkaloid (-)- Morphine Hydrochloride (-)-Codeine Phosphate (-)-Morphine Hydrochloride . 17 CH3 . HCl 3H O 2 N 10 9 11 D 14 8 1 B 7 A C 2 E 6 HO 3 4 O 5 OH (-)-Morphine Hydrochloride 17 H N 10 9 11 14 8 1 7 2 6 3 4 5 morphinan (5α,6α)-7,8-didehydro-4,5-epoxy-17-methylmorphinan-3,6-diol hydrochloride trihydrate The Structure of (-)-Morphine 17 CH3 10 N 9 D 14 8 1 11 B 7 A C 2 E 6 HO 3 4 O 5 OH Morphine is composed of five fused rings, and the molecule has five chiral centers with absolute stereochemistry (5R, 6S, 9R, 13S and 14R). The naturally occurring isomer of morphine is levo [(l) or ()] rotatory. (+)-morphine has been synthesized, and it is devoid of analgesic and other opioid activities. The configuration of (-)-morphine H H N H3C D H H OH C B E O A OH B/C and C/E are cis, and C/D is trans. Morphine is shaped like a three-dimensional "T" with rings A, B, and E forming a near perfect vertical plane and rings C and D forming a more distorted horizontal plane. The D ring is in the chair conformation and the C ring is a boat with atoms 6 and 14 fore and aft. The "T" is skewed. 吗啡的理化性质 1. 吗啡是从阿片中提取得到的。 2. 吗啡显弱酸性, 可与强碱成盐。 3. 吗啡显碱性,可与酸成盐,临床上常用其盐酸 盐或硫酸盐。 4. 吗啡具有左旋性。 5. 吗啡及其盐易被氧化。 Oxidation of (-)-morphine O N CH3 N CH3 N CH3 N CH3 + HO O OH HO O OH OH O Pseudomorphine or Dimorphine OH HO O OH morphine N-oxide Its stability is related with pH of solution. it is the most stable in the solution (pH=4). 在中性或碱性条件下极易氧化,在日光、重金属离子存在 下可催化此反应,所以盐酸吗啡注射液放置过久颜色变深 ,在制备制剂及贮存时应加以注意。 Identification of apomorphine in morphine CH3 N CH3 N HCl or H3PO4 HO O OH Apomorphine HO OH 催吐剂 CH3 HN I2 NaHCO3 O O 醚层显宝石红 水层显绿色 Identification of little morphine in codeine (1) N CH3 N CH3 NaNO2 NO HCl HO O OH O OH HO 2-nitrosomorphine NH4OH marron(ºì ×ØÉ«) N CH3 NaNO2 CH3O O OH HCl Identification of morphine in codeine (2) A. 水溶液与三氯化铁试液反应显蓝色(酚羟基)。 B.水溶液加入稀铁氰化钾试液后再与三氯化铁试 液反应,生成蓝色(酚羟基) 。 C17H19O3N+K3Fe(CN)6 → C34H36O6N2 C34H36O6N2+ K4Fe(CN)6 + FeCl3 → blue Metabolism of morphine H H N+ CH3 N+ CH3 first pass effect morphine O HO OH O RO R=Glu or SO3- CYP450 N-demethylation H H + N N+ CH3 H activity Nomorphine HO OH O OH HO O O-Glu The use and action in the body of morphine It is available in intramuscular, subcutaneous(皮 下的), oral, rectal, epidural(硬膜外的) and intrathecal(鞘内的) dosage forms. Morphine is three to six times more potent when given intramuscularly than it is given orally. The difference in activity is due to extensive firstpass 3-O-glucuronidation of morphine -- an inactive metabolite. its bioavailability is about 25%. Pharmacologic activity of (-)- morphine (-)-Morphine, which is a μopioid agonist, is the natural product prototype for this class of analgesics. Morphine hydrochloride or sulfate is the most often used analgesic for severe, acute, and chronic pain. Overdoses of morphine as well as all μ agonists in this section can be effectively reversed with naloxone (盐酸 纳洛酮) . Side effects of (-)-morphine Sedation(镇静), dizzy(眩晕), vomit(呕吐), drowsiness(嗜睡), constipation(便秘) Tolerance, dependence Respiratory depression, which is the cause of death from opioid overdose. (-)-Codeine Phosphate CH3 N . H3PO4 . 1.5H2O CH3O O OH (5α,6α)-7,8-didehydro-4,5-epoxy-3-methoxy-17methylmorphinan-6-ol phosphate sesquihydrate Synthesis of Codeine N CH3 N CH3 C6H5N+(CH3)3OH- HO O morphine OH CH3O O codeine OH 可待因的理化性质 可待因具有左旋性。 较吗啡稳定,但遇光仍易变质。 水溶液加氨试液后无沉淀。 与三氯化铁试液反应不显色。 Stability of Codeine protection from light CH3 N CH3O O OH It is more stable than morphine. Identification of little morphine in codeine N CH3 N CH3 NaNO2 NO HO O OH HCl O OH HO 2-nitrosomorphine NH4OH marron(ºì ×ØÉ«) N CH3 NaNO2 CH3O O OH HCl Metabolism of codeine H H H N+ CH3 N+ CH3 N+ CH3 10% CYP450 O-Demethylation OH HO O CH3O CYP450 Codeine N-demethylation OH N H O + N H OH HO OH R=Glu or SO3- H + O RO CYP450 morphine N-demethylation H CH3O O O N+ CH3 H OH nomorphine HO O O-Glu Pharmacologic activity Codeine is a weak μ agonist, which has about 1/10 analgesic potency of morphine. Codeine phosphate is used extensively to treat moderate-to-mild pain and it is a more effective antitussive (止咳剂) agent than morphine. Codeine causes less sedation, nausea(恶心), vomiting, constipation, tolerance, dependence and has less effect on the gastrointestinal and urinary tracts than morphine. (2) Structural modification of morphine CH3 N HO O H OH SARs for morphine derivatives Drugs Substituent Activity Codeine(可待因) 3-OCH3 10×decrease Ethylmorphine(乙基吗啡) 3-OCH2CH3 10×decrease Heteromorphine(异可待因) 6-OCH3 Heroin(海洛因) 3,6-diOAc 10×increase (toxicity) increase Hydromorphone(氢吗啡酮) 6-keto, 7,8-dihydro 10×increase Oxymorphone( 羟吗啡酮) 6-keto, 7,8-dihydro, 14β-OH 10×increase Hydrocodone(氢可酮) 3-OCH3, 6-keto, 7,8-dihydro, decrease Oxycodne(羟考酮) 3-OCH3, 6-keto, 7,8-dihydro,14-OH decrease N-2-phenylethyl-morphine N-β-苯乙基吗啡 NCH2CH2Ph 14×increase Nalorphine(烯丙吗啡) NCH2CH=CH2 μ antagonist Importance to structural modification of morphine In the structure of morphine (or any other opioid) are likely to cause a change in the affinity and intrinsic activity of the new compound for each of the opioid receptor types, i.e., a selective μ agonist may shift to become a selective К agonist. In addition, the new compound has different physical properties than its parent. The different physical properties (e.g. solubility, partition coefficient, pKa) result in different pharmacokinetic characteristics for the new drug and can affect its in vivo activity profile. Heroin CH3 N AcO O H OAc The 3,6-diacetyl derivative of morphine is known commonly as heroin. Heroin was synthesize from morphine in 1874 and was introduced to the market in 1898 by the friedrich Bayer Co. in Germany. The reason that heroin has higher activities Heroin itself has relatively low affinity for μ opioid receptors; however, its high lipophilicity compared with morphine results in enhanced penetration of the blood-brain barrier. Once in the body, esterases hydrolyze the 3acetyl group to produce 6-acetyl-morphine. This latter compound has μ agonist activity in excess of morphine. The reason that heroin is limited Heroin provides an “euphoric rush” that makes this compound a popular drug of abuse. Repeated use of heroin results in the development of tolerance, physical dependence that is often destructive to the user and to society. In addition, the use of unclean or shared hypodermic (皮下) needles for self-administering heroin often results in the transmission of AIDs, hepatitis, and other infections. (3) Synthetic Analgesics A. Morphinans (吗啡喃类) B. Benzomorphans(苯吗喃类) C. Piperidines(哌啶类) D. Phenylpropylamines(苯丙胺类) E. Aminotetralins(氨基四氢萘类) F. Cyclohexane derivatives(环己烷衍生物 ) A. Morphinans(吗啡喃类) CH3 N R' N H D A HO O morphine OH B C R morphinan Removal of 3,4-epoxide bridge in the morphine structure results in compounds that are referred to as morphinans. The synthetic produce yields compounds as racemic mixtures, but only the levo (-) isomers possess opioid activity. The dextro isomers have useful antitussive activity. Levophanol(左啡诺) CH3 N COOH . H HO C OH C H . 2 H2O COOH HO 17-methylmorphinan-3-ol tartrate dihydrate Levophanol is about 8 times more potent as an analgesic in humans than morphine. Levophanol’s increased activity is due to an increase in affinity for μ opioid receptors and its greater lipophilicity, which allows higher peak concentrations to reach the brain. The configuration of levophanol H H H3C N C D B levophanol A OH H H N H3C D H H OH C B E O A OH morphine Butophanol(布托啡诺) CH2 N OH HO Butorphanol is a μ antagonist and К agonist. The mixed agonist / antagonist is an antagonist analgesic and has less dependence liability. B. Benzomorphans (苯吗喃类) R' N D R B A R HO Synthetic compounds that lack both the epoxide ring and the C ring of morphine retain opioid activity. Compounds having only the A,B, and D rings are named chemically as derivatives of 6,7-benzomorphan or benzomorphans. Pentazocine (喷他佐辛) CH2CH=C(CH3)2 N CH3 CH3 HO Pentazocine has an agonist action on κ opioid receptors--an effect that produces analgesia, and pentazocine is weak antagonist at μ receptors. Pentazocine is a mixed agonist/antagonist and has less dependence. C. Piperidine (哌啶类) 4-phenylperidines (4-苯基哌啶类) 4-anilidopiperidines (4-苯胺基哌啶类) Pethide(哌替啶) C6H5 COOC2H5 N CH3 The analgesic activity of pethide was discovered during biologic screening of compounds that were designed as anticholinergics(抗胆碱能类) patterned on the structure of atropine. Pethide is a typical μ agonist with 1/6~1/8 the potency of morphine. The conformation of pethidine O N H3C D H H N H3C D C£-O£-C2H5 H H OH C B A E O A OH N H3C D A COOC2H5 Analgesic compounds in the 4-Phenylpiperidine class may be viewed as A, D ring analogs of morphine. It created great interest because the structural simplicity of the drug and the synthesis of fragments of the morphine molecule to provide a nonaddictive and potent analgesic was futile. Alphaprodine(阿法罗定) O C6H5 COC2H5 O C6H5 OCC2H5 CH3 N CH3 N CH3 α- prodine is the 3-methyl reversed ester derivatives of pethidine. α- prodine is as active as morphine. It has a more rapid onset and shorter duration of action than morphine. Fentanyl(芬太尼) C6H5 OCOC2H5 C6H5 N COC2H5 CH3 N CH3 N CH2CH2C6H5 Structural modification of the 4-phenylpiperidines has led to discovery of the 4-anilidopiperidine or the fentanyl group of analgesics. Fentanyl and its derivatives are μ agonists, and they produce typical morphine-like analgesia and side effects. Structural variations of fentanyl Structural variations of fentanyl that have yielded active compounds are substitution of an isosteric ring of the phenyl group, addition of a small oxygen-containing group at the 4-position of the piperidine ring. Newer drugs that illustrate some of these structural changes are alfentanil(阿芬他尼) and sulfentanil(舒芬太尼). Both of these drugs have higher safety margins (安全范围) than other μ agonists. They have more rapid onset and shorter duration of action than morphine. Sulfentanil(舒芬太尼) C6H5 COC2H5 N CH2OCH3 N CH2CH2 Bioisosterism S Alfentanil(阿芬太尼) C6H5 COC2H5 N CH2OCH3 O N CH2CH2 CH2CH3 N N Bioisosterism N N Refentanil(瑞芬太尼) C6H5 COC2H5 N R1=±½»ù£¬ R2=H ·ÒÌ« Äá R1=±½»ù£¬ R2=COOCH3¿¨ ·ÒÌ« Äá R N CH2CH2R1 C6H5 COC2H5 N R N CH2CH2COOH ÎÞ »î ÐỐú лÎï Èí Ò© Éè¼Æ C6H5 COC2H5 N CO2CH3 N CH2CH2COOCH3 Èð·ÒÌ« Äá t1/2=10~21min ²» »á Ðî»ýÖж¾£¬¼õÉÙºô Îü ÒÖÖÆ µÄΣ ÏÕ Ohmefentanyl(羟甲芬太尼) C6H5 C6H5 N COC2H5 N COC2H5 CH3 8 N N C6H5 C 6 H5 ·ÒÌ« Äá(Fentanyl) OH 6000 ôǼ׷ÒÌ« Äᣨ Ohmefentanyl£© 13000 Pethidine Hydrochloride (Dolantin) C6H5 COOC2H5 . HCl N CH3 1-methyl-4-phenyl-4-piperidinecarboxylic ethyl ester hydrochloride Synthesis of pethidine CH2CN + CH3N(CH2CH2Cl)2 NaNH2 N CH3 NC H2SO4 C2H5OH N CH3 N HOOC C2H5OOC HCl N CH3 . HCl C2H5OOC II I III CH3 Physical-chemical properties N N H3C CO2C2H5 H3C CO2C2H5 . HCl + Na2CO3 white crystalline power oil solid mp 30~31¡æ Hydrolysis N H3C CO2C2H5 H+ short time Identifying reaction OH C6H5 CO2C2H5 O2N NO2 C6H5 ON2 NO2 . + N N CH3 OH CO2C2H5 NO2 CH3 NO2 Yellow Mp 188~191℃ Metabolism of pethidine C6H5 CO2C2H5 C6H5 CO2H C6H5 CO2-Glu N N N CH3 CH3 CH3 Pethidine acid C6H5 CO2C2H5 N H Norpethide (no activity,toxity) C6H5 CO2H N H Norpethidinic acid no activity C6H5 CO2-Glu N H Pharmacologic activity (1) Pethidine is a μ agonist with about 1/10 the potency of morphine after intramuscular dose. 主要用于创伤、手术后或癌症晚期的疼痛。 Pethidine produces the analgesia, dependence, respiratory depression, and euphoria caused by other μ agonists, but it causes less constipation, and it does not inhibit cough. When given orally, pethidine has 40~60% bioavailability as a result of significant first-pass metabolism. Pharmacologic activity (2) Pethidine has received extensive use in obstetrics( 产科学) because of its rapid onset, short duration of action. Prolonged dosage of pethidine may cause an accumulation of the metabolite normeperidine. Normeperidine has only weak analgesic activity, but it causes CNS excitation. Pethidine has a strong adverse reaction when given to the patients receiving a monoamine oxidase (MAO) inhibitor. Fentanyl Citrate (枸橼酸芬太尼) CH2COOH CH3CH2CO - N C6H5 N CH2CH2C6H5 HO - C COOH CH2COOH N-Phenyl-N-[1-(2-phenylethyl)-4-piperidinyl] propanamide citrate Design of synthesis routine CH2COOH CH3CH2CO - N N CH2CH2C6H5 HO - C COOH CH2COOH C6H5 III II I IV Synthesis of Fentanyl Citrate (1) CH2=CHCO2CH3 CH2CH2NH2 CH3ONa CH2CH2N CH2CH2N O CO2CH3 CH2CH2N O Ph-NH2 piperidine CH2CH2N N CH2CH2CO2CH3 CH2CH2CO2CH3 HCl Synthesis of Fentanyl Citrate (2) H2/Ni CH2CH2N N H COCH2CH3 (CH3CH2CO)2O CH2CH2N N COCH2CH3 citric acid CH2CH2N N CH2COOH HO - C COOH CH2COOH Pharmacologic activity Fentanyl is a μ agonist with about 80 times the poyency of morphine. The advantages of fentanyl over morphine for anesthetic procedures are its short duration of action (1to 2 hours). A fentenyl patch has been released for the treatment of severe chronic pain. Fentanyl’s short duration of action after parenteral dosing is due to redistribution, rather than to metabolism or excreceion. Repeated doses of fentanyl can result in accumulation and toxicities. Elderly patients are usually more sensitive to fentanyl and require lower doses. Sulfentanil Citrate(枸橼酸舒芬太尼) C6H5 N COC2H5 CH2OCH3 CH2COOH HO - C COOH N CH2CH2 S CH2COOH Addition of the 4-methoxymethyl group and bioisosteric replacement of the phenyl with a thiophenyl on the fentenyl structure results in a 10-fold increase in μ opioid activity. Pharmacologic activity Sufentanil is 600 to 800 times more potent than morphine. Despite its greater sedative and analgesic potency, sufentanil produces less respiratory depression at effective anesthetic doses. Sufentanil is available in an intravenous dosage form, and it is used for anesthetic procedures. It has a faster onset and shorter duration of action than fentenyl. The shorter duration of action is due to redistribution from brain tissues. D. Phenylpropylamine German scientists synthesized another series of open chain compounds as potential antispasmodics(解痉药). Methadone was the major drug to come from this series of compounds. (+)-Methadone CH3 O C2H5 H3C N H3C Methadone is especially useful for its oral activity and its long duration of action. These properties make methadone useful for maintenance therapy of opioid addicts and for pain suppression in the terminally ill (e.g., hospice临终关怀医院programs). Methadone It has moderate dependence liability and constipating(便秘) effects. Methadone is a respiratory depressant and cough suppressant. The withdrawal syndrome is slower in onset, more protracted(延长), and less severe than that seen with heroin. Accordingly, it is used to detoxify dependent subjects. Because of its relatively mild sedative and euphoric effects, it is also used for maintenance during rehabilitation (康复). The configuration of Methadone H3C C2H5 CH3 N: N H3C O A D ¦Ä+ COOC2H5 H3C pethidine A CH3 N D H B C A E HO O morphine OH Dextropropoxyphene (右丙氧芬) CH3 H3C N H3C O COCH2CH3 CH2 Propoxyphene is an open chain compound that was discovered by structural variation of methadone. Propoxyphene is a weak μopioid agonist, having only one-fifteen the activity of morphine. The active (+)isomer has (2S,3R) absolute configuration. Methadone Hydrochloride H3C N HCl . H3C 7 CH3 O 5 C2H5 3 4 6 6-(dimethylamino)-4,4-diphenyl-3-heptanone hydrochloride Synthesis of methadone N C NaNH2 N H3C C + N(CH3)2 + Cl H3C O H3C (1) C2H5MgBr + + C CH3 (2) H H3C methadone N(CH3)2 N(CH3)2 isomethadone C CH3 N(CH3)2 O H3C N N(CH3)2 Reduction reaction CH3 O H3C N H3C C2H5 . (i-PrO)3Al or Na-Hg Identification of methadone (1) CH3 O H3C N H3C C2H5 NH2NHCONH2 Identification of methadone (2) H3C CH3 O C2H5 N H3C H3C CH3O C2H5 N H3C .HO3S N N + HO3S N N ¼×»ù³È N(CH3)2 yellow N(CH3)2 The metabolism of methadone The elimination of methadone depends on liver function and urinary pH. The typical half-life is 19 hours, but if urinary pH is raised from normal values of 5.2 to 7.8, the half-life becomes 42 hours. At the higher pH, a lower percentage of methadone exists in the ionized form, and there is more renal reabsorption of the drug. Enzyme inducer (e.g., phenytoin, rifampin) can lead to withdrawal in patients using methadone for maintenance of addiction. Toxic doses can build up in patients with liver disease or in geriatric patients(老年病人) who have a decreased oxidative metabolism capacity. Metabolism of methadone in the liver CH3 N CH3 CPY-450 CH3 O CH3 N H O N CH3 HO Methadol activity CH3 no activity alcohol dehydrogenase CH3 N CH3 CPY-450 CH3 CH3 CH3 NH HO Normethadol activity CH3 NH2 CPY-450 HO CH3 Dinormethadol activity greater oral potency and longer duration of action Pharmacologic activity(1) Methadone is a synthetic agent with about the same μ opioid potency as morphine. The drug is used as a racemic mixture, but nearly all of the activity is due to the (-)-isomer. Methadone’s usefulness is a result of its greater oral potency and longer duration of action compared with other μ agonists. Pharmacologic activity(2) Methadone is an excellent analgic (无痛感的) for use in cancer patients, and it is often used in hospice programs. Oral doses of 40mg are commonly used for 24 hour suppression of withdrawal symptoms (addiction maintainence) in opioid addicts. Methadone requires once-a-day dosing, usually at a clinic, to suppress withdrawal symptoms. α- Methadol( LAAM ) Although methadone is a good drug for maintenance of addiction, it is not deal. Methadone requires oncea-day dosing, usually at a clinic, to suppress withdrawal symptoms. Once-a-day dosing is expensive and sometimes logistically difficult to achieve. LAAM is available and is used in some treatment programs to overcome the problems of methadone. LAAM is more potent than methadone, and it has a longer duration of action. A single oral dose of this agent can suppress abstinence withdraw for up to 3 days. E. Aminotetralin(氨基四氢萘) Aminotetralins represent A, B ring analogs of morphine. A number of active compounds in this class have been described, but only dezocine (地左辛), a mixed agonist/antagonist, has been marketed. Dezocine (地左辛) CH3 N H3C D B NH2 HO A H C A B E HO O OH morphine Dezocine possess agonist/antagonist effects (antagonist analgesics), and has less dependence liability. Tramadol(曲马朵) CH3 N A D OH CH2N(CH3)2 CH3O H B C A E C HO O OH morphine Tramadol is weak μ receptor agonist. 对呼吸抑制作用小,短时间应用时成瘾性小,用 于中重度疼痛的止痛。 (4)High potent μagonists Etorphine(埃托啡) Dihydroetorphine(二氢埃托啡) Etorphine(埃托啡) N CH3 HO HO O C3H7-n CH3 OCH3 Etorphine is about 1000 times more potent than morphine as μ agonist. Etorphine has a low therapeutic index in humans, and its respiratory depressant action is difficult to reverse with an opioid antagonists; thus, the compound is not useful in medical practice. Dihydroetorphine(二氢埃托啡) N CH3 HO HO O C3H7-n CH3 OCH3 二氢埃托啡的镇痛作用强于埃托啡,动物实验结果显示戒 断症状及精神依赖性均明显轻于吗啡。 二氢埃托啡1991年上市,开始作为非麻醉品用于临床, 但在使用中发现有较强的精神依赖性和躯体依赖性,耐受 性形成很快,成瘾性强,滥用潜力大,1992年开始按麻 醉药品管理。 Section 2 Opioid Antagonists and agonist / antagonist If the N-methyl of rigid opioid analgesics (such as morphine, morphinans and benzomorphans) is substited by allyl, cyclopropylmethyl (CPM) or cyclobutylmethyl (CBM), these analgesics become μ antagonists, but they are also agonists of other opioid receptor subtype, such as κ receptor. (1) Opioid Antagonists Nalorphine (烯丙吗啡), naloxone (纳洛酮) and naltrexone (纳曲酮) are clinically used to detoxify opioid analgesics. Nalorphine(烯丙吗啡) N CH2CH=CH2 HO O OH Nalorphine is an opioid agonists/antagonists. It is a μ antagonist and κ receptor agonist. 严重的焦虑、致幻等精神症状,不能作为镇痛药。 Naloxone(纳洛酮) N CH2CH=CH2 OH HO O O Naloxone is a “pure” opioid antagonist. 因有首过效应,纳洛酮须非肠道给药。 Naltrexone(纳曲酮) N CH2 OH HO O O Naltrexone is a “pure” opioid antagonist. 纳曲酮与纳洛酮比较,作用强,口服,维持时间长。 (2) Opoid agonists/antagonists 喷他佐新于1976年进入临床,成为第一个拮抗性 镇痛药。 其他有布托啡诺、丁丙诺啡和地左辛。 Pentazocine(喷他佐辛) 10 3 CH2CH=C(CH3)2 1 N 2 11 4 CH3 5 9 HO 8 7 6 CH3 (2α, 6α,11R)-1,2,3,4,5,6-hexahydro-6,11-dimethyl-3-(3methyl-2-butenyl)-2,6- methano-3-benzazocin-8-ol, Talwin 喷他佐辛属于苯吗喃类,为混合的激动-拮抗剂,即为μ受体 弱拮抗剂(纳洛酮的1/30),κ受体激动剂(镇痛作用是吗 啡的1/6),不良反应较小,成瘾性小,为非麻醉药品。 喷他佐辛口服有首过效应,临床用于减轻中度至重度疼痛。 Butophanol(布托啡诺) CH2 N OH HO Butorphanol is a μ antagonist (about 1/6 the potency of naloxone) and strong κ agonist (5 times more potent than morphine). The mixed agonist / antagonist has less dependence liability. 缺点:首过效应, 镇静、焦虑等κ受体激动剂副作用,心 血管副作用。 临床用于中度至重度疼痛止痛和辅助麻醉。 Nalbuphine hydrochloride (盐酸纳布啡) N CH2 OH .HCl HO O OH Nalbuphine is a μ antagonist (about 1/4 the potency of naloxone) and strong κ agonist (about the same potency as morphine). The antagonist analgesic has less dependence liability. 缺点:首过效应, κ受体激动剂副作用。 临床用于中度至重度疼痛止痛和辅助麻醉。 Buprenorphine Hydrochloride (盐酸丁丙诺啡) N CH2 . HCl CH3 C(CH3)3 HO O OH OCH3 盐酸丁丙诺啡为μ受体强的部分激动剂,κ受体部分激动剂 和δ受体拮抗剂。对阿片受体有高亲脂性,镇痛作用强于吗 啡(20~50倍),但不能被纳洛酮拮抗。 呼吸抑制副作用轻,单独用耐受性和成瘾性小,临床用于中 度至重度疼痛止痛和海洛因戒毒。 Dezocine (地左辛) H3C NH2 HO A B Dezocine possess agonist/antagonist effects (antagonist analgesics), and has less dependence liability. Section 3 Endogenous opioid peptides 1973年Simon, Snyder and Terenius证实鼠脑内 存在立体特异性的阿片样镇痛药的结合位点,这 些位点存在于包括人和所有脊椎动物的中枢神经 系统及一些外周平滑肌系统的神经组织中。 体内既然存在阿片受体,必然有尚未鉴定的内源 性配体存在。 Endogenous opioid peptides 1. 1975年Hughes and Kosterlitz等从猪脑内提取 、分离、纯化、鉴定得到两个吗啡样镇痛作用多 肽:Enkephalin(脑啡肽): A. methionine enkephalin (Tyr-Gly-Gly-Phe-Met) (甲硫氨酸脑啡肽, Met-E) B. leucine enkephalin (亮氨酸脑啡肽, Leu-E) (Tyr-Gly-Gly-Phe-Leu) 。 All these peptides are called endorphin(内啡肽) or opioid peptides(内源性阿片样肽类) 2.β-endorphin (β-内啡肽, 31peptides): Tyr-Gly-GlyPhe-Met-Thr-Ser-Glu-Lys-Ser-Gln-Thr-Pro-Leu-ValThr-Leu-Phe-Lys-Asn-Ala-Ile-Ile-Lys-Asn-Ala-TyrLys-Lys-Gly-Glu. dynorphin 1-17 (强啡肽, 17 peptides ): Tyr-GlyGly-Phe-Leu-Arg-Arg-Ile-Arg-Pro-Lys-Leu-Lys-TrpAps-Asn-Gln. α-neoendorphin(α-新内啡肽,10 peptides ): Tyr-Gly-Gly-Phe-Leu-Arg-Lys-Tyr-Pro-Lys. β-neoendorphin(β-新内啡肽,9 peptides ):Tyr-Gly-Gly-Phe-Leu-Arg-Lys-Tyr-Pro. Section 4 Structure-Activity Relationships of Opioid Analgesics μ Receptor Agonists κ Receptor Agonists δ Receptor Agonists 1.Structure-Activity Relationships of μ Receptor Agonists 1. 吗啡为μ受体选择性激动剂,左旋体有活性。 2. μ受体激动剂结构中均有A环和碱性叔胺N原子,是镇痛作 用必需的。 在生理pH条件下,叔胺N原子形成正离子。 3. A环与碱性N原子或者通过2个碳原子的碳链(相当于B环的 9、10位)或者通过3个碳原子的碳链(经过哌啶环的一边) 相连接。 4. 刚性结构μ受体激动剂 3-酚羟基使活性显著增强。 5. 叔胺N原子上取代基的大小对具有激动或拮抗活性有重要 影响。 6. 哌啶类和氨基酮类柔性结构和吗啡等刚性结构具有共同的 药效构象。 柔性结构和刚性结构具有共同的药效构象 H H N H3C D H H OH H3C C B E H H C2H5 N H3C C D CH3 O N: ¦Ä+ B O A A H3C OH OH A H H N+ O N H3C D D C£-O£-C2H5 B A A OH 2. Structure-Activity Relationships of κ Receptor Agonists All of the market κ agonists have structures related to the rigid opioids and N-ally, N-CPM, or N-CBM substitutions. The compounds are all μ receptor antagonists and κ receptor agonists. 吗啡类、吗啡喃类和苯吗喃类等刚性结构中的N 上被allyl、CPM或CBM取代,这些化合物具有激 动和拮抗双重活性。 The κ agonist activity is enhanced if there is an oxygen group placed at the 8-position (e.g., ethylketazocine, 乙基酮佐新). The oxygen group in an N- substituent also enhances κ activity. Ethylketazocine (乙基酮佐新) O N 8 CH3 HO CH3 在6,7-苯并吗啡烷环上引入含氧基团可使κ 阿片受体激动活性增强。 Bremazocine(布马佐新) OH N CH3 CH3 HO CH3 在6,7-苯并吗啡烷环N-取代基上引入含氧基团 可使κ阿片受体激动活性增强。 3. Structure-Activity Relationships of δ Receptor Agonists Structure-activity relationships for δ receptor agonists are the least developed among the opioid compounds. Nonpeptide δ selective agonists are just beginning to be discovered. Peptides with high selectivity for δ receptors are known. The SARs for some of these peptides are discussed in the following. 4.Structure-Activity Relationships of Endogenous Opioid Derivatives 1. All of the endogenous peptides have Leu- or Metenkephalin as their first amino acid residues. 2. The tyrosine at the first amino acid residue position of all the endogenous opioid peptides is essential for activity. Removal of the phenolic hydroxyl group or the basic nitrogen abolishes activity. The Tyr1 free amino group may be alkylated (methyl or ally groups to gives agonists and antagonists), but it must retain its basic character. The structure resemblance between morphine and the Tyr1 group of opioid peptides is especially obvious. 3.The next most important moiety in the enkephalin structure is phenyl group of Phe4. Removal of this group or changing its instance from Try1 can result in full of substantial loss in activity. 4. The replacement of the natural L-amino acids with unnatural D-amino acids can make the peptides resistant to the actions of several peptidases that generally rapidly degrade the natural endorphins. The placement of bulky groups into the structure (e.g., the addition of N-Me to Phe4) also slows the action of peptidases. Conversion of the terminal carboxyl group into an alcohol or an amide protects the compound from carboxy peptidases. 5. Structural changes that highly restrict the formational mobility of the peptides (e.g., substitution of proline for Gly2 or cyclization of the peptide) have been especially useful for the discovery of receptor selective opioid peptides. 脑啡肽分子内形成氢键 O H O H N N H H H N O O H N COOH N O H CH3 CH3 5. Models of the Opioid Receptor 根据吗啡和合成镇痛药的共同的药效构象,1954 年,Beckett和Casy提出的阿片受体模型。 Beckett和Casy提出的阿片受体模型 负离子部位 - 凹槽 H H3C H N H H OH O 适合芳环的平坦区 OH 氢键接受部位 三点论 μ阿片受体激动剂具有以下结构 具有一个碱性中心; 具有一个平面芳环结构, 碱性中心与芳环几乎共平 面; 烃基链部分凸出于平面。 埃托啡及其衍生物与μ阿片受体结合图形 OH OCH3 HO O H3C N+ 负离子部位 凹槽 H CH3 - 适合芳环的平坦区 氢键接受部位 A 四点论 刚性结构和柔性结构的吗啡样化合物N-取代基和芳 环部分可能结合在受体不同亚部位 Portoghese 注意到刚性结构的吗啡、吗啡喃或苯吗喃类, 当N-取代基的改变时,镇痛作用相应改变,说明刚性结构的 吗啡样化合物以相同的方式与受体结合呈现镇痛活性。 柔性结构哌替啶等,N-取代基的改变,不能产生刚性结构的 吗啡样化合物那样相应的活性改变。因此认为刚性的和非刚 性的阿片样镇痛药结构中N-取代基部分,分别结合在受体的 不同表面上,而呈现不同的镇痛性。 后来发现刚性的吗啡等镇痛药,当芳环上有3-OH取代,活性 增强,而在非刚性系列,例如哌替啶衍生物烯丙基罗定,当 其结构中的间位被OH取代,镇痛作用消失。因此认为,这两 个系列中的芳环结构部分,分别结合在受体表面分离的不同 亚部位。 Allyprodine(烯丙基罗定) OCOC2H5 R N CH3 H H3C H N H H OH O OH morphine 脑啡肽与μ阿片受体相互作用 A O R G OH N+ N NH M O N O O Tyr-Gly-Gly-Phe P T 吗啡与μ阿片受体相互作用 G OH A P O H3O N+ H OH T 哌替啶衍生物与μ阿片受体相互作用 A H N+ CH3 O R O P, 疏水性 CH3 T 东罂粟碱与μ阿片受体相互作用 H3CO H3C P OH O T OH H3C NH+ A N CH3 HO HO O CH3 OCH3 Summary 1. The classifying of synthesis analgesics Piperidines: Pethidine, Fentanyl; Phenylpropylamines: Methadone; Morphinans: Levorphanol; Benzomorphans: Pentazocine; Aminotetralins: Dezocine Cyclohexane derivatives: Tramadol. 2. Opioid anatagonists and antagonist analgesics Opioid anatagonists(Nalorphine, Naloxone and Naltrexone) : pharmacologic action and chemical structure characteristics. Antagonist analgesics(pentazocine): pharmacologic action and chemical structure characteristics. 3. Structure-activity relationships of μreceptor agonists μ激动剂化学结构中均有A环和碱性氮原子。 A环与碱性氮原子或者通过2个碳原子的碳链(相当 于B环的9、10位)或者通过3个碳原子的碳链(经 过哌啶环的一边)相连接。 3-酚羟基使活性显著增强。 叔胺结构对镇痛作用是必需的,N原子上取代基的 大小对具有激动或拮抗活性有重要影响。 4. Models of the opoid receptor μ阿片受体激动剂的结构特点: 具有一个碱性中心; 具有一个平面芳环结构; 碱性中心与芳环几乎共平面; 烃基链部分凸出于平面。 5. Clinically available agents Morphine Hydrochloride: chemical structure characteristics, physical-chemical properties and pharmacologic action. Pethidine Hydrochloride: chemical structure characteristics and pharmacologic action. Methadone Hydrochloride: chemical structure characteristics and pharmacologic action. Fentanyl Citrate: chemical structure characteristics, pharmacologic action and synthesis routine.