Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Interactome wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Endogenous retrovirus wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Point mutation wikipedia , lookup
Enzyme inhibitor wikipedia , lookup
Peptide synthesis wikipedia , lookup
Biosynthesis wikipedia , lookup
Signal transduction wikipedia , lookup
Western blot wikipedia , lookup
Metalloprotein wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Protein–protein interaction wikipedia , lookup
Two-hybrid screening wikipedia , lookup
Biochemistry wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
Polyclonal B cell response wikipedia , lookup
Proteolysis wikipedia , lookup
Clinical neurochemistry wikipedia , lookup
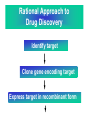
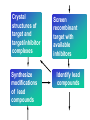
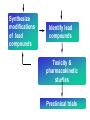
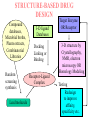
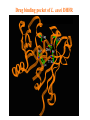
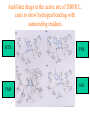
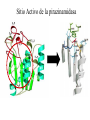
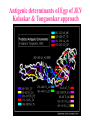
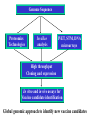
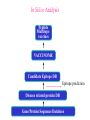
Use of bioinformatics in drug development and diagnostics Bringing a New Drug to Market Review and approval by Food & Drug Administration 1 compound approved Phase III: Confirms effectiveness and monitors adverse reactions from long-term use in 1,000 to 5,000 patient volunteers. Phase II: Assesses effectiveness and looks for side effects in 100 to 500 patient volunteers. 5 compounds enter clinical trials Phase I: Evaluates safety and dosage in 20 to 100 healthy human volunteers. 5,000 compounds evaluated 0 2 4 6 8 Discovery and preclininal testing: Compounds are identified and evaluated in laboratory and animal studies for safety, biological activity, and formulation. 10 Source: Tufts Center for the Study of Drug Development 12 14 Years 16 Biological Research in 21st Century “ The new paradigm, now emerging is that all the 'genes' will be known (in the sense of being resident in databases available electronically), and that the starting "point of a biological investigation will be theoretical.” - Walter Gilbert Rational Approach to Drug Discovery Identify target Clone gene encoding target Express target in recombinant form Crystal structures of target and target/inhibitor complexes Synthesize modifications of lead compounds Screen recombinant target with available inhibitors Identify lead compounds Synthesize modifications of lead compounds Identify lead compounds Toxicity & pharmacokinetic studies Preclinical trials An Ideal Target • Is generally an enzyme/receptor in a pathway and its inhibition leads to either killing a pathogenic organism (Malarial Parasite) or to modify some aspects of metabolism of body that is functioning dormally. • An ideal target… – – – – – Is essential for the survival of the organism. Located at a critical step in the metabolic pathway. Makes the organism vulnerable. Concentration of target gene product is low. The enzyme amenable for simple HTS assays How Bioinformatics can help in Target Identification? • • • • • Homologous & Orthologous genes Gene Order Gene Clusters Molecular Pathways & Wire diagrams Gene Ontology Identification of Unique Genes of Parasite as potential drug target. Comparative Genomics Malarial Parasites: Source for identification of new target molecules. • Genome comparisons of malarial parasites of human. • Genome comparisons of malarial parasites of human and rodent. • Comparison of genomes of – – Human – Malarial parasite – Mosquito What one should look for? Human P.f Mosquito Proteins that are shared by – •All genomes •Exclusively by Human & P.f. •Exclusively by Human & Mosquito •Exclusively by P.f. & Mosquito Unique proteins in – Human P.f. Targets for anti-malarial drugs Impact of Structural Genomics on Drug Discovery Dry, S. et. al. (2000) Nat. Struc.Biol. 7:976-949. Drug Development Flowchart • Check if structure is known • If unknown, model it using KNOWLEDGE-BASED HOMOLOGY MODELING APPROACH. • Search for small molecules/ inhibitors • Structure-based Drug Design • Drug-Protein Interactions • Docking Why Modeling? • Experimental determination of structure is still a time consuming and expensive process. • Number of known sequences are more than number of known structures. • Structure information is essential in understanding function. Sequence identities & Molecular Modeling methods Methods Sequence Identity with known structures • ab initio 0-20% • Fold recognition 20-35% • Homology Modeling >35% STRUCTURE-BASED DRUG DESIGN Compound databases, Microbial broths, Plants extracts, Combinatorial Libraries Random screening synthesis Lead molecule 3-D ligand Databases Docking Linking or Binding Receptor-Ligand Complex Target Enzyme OR Receptor 3-D structure by Crystallography, NMR, electron microscopy OR Homology Modeling Testing Redesign to improve affinity, specificity etc. Binding Site Analysis • In the absence of a structure of Targetligand complex, it is not a trivial exercise to locate the binding site!!! • This is followed by Lead optimization. Lead Optimization Active site Lead Lead Optimization Compounds which are weak inhibitors may be modified by combinatorial chemistry in silico if the target structure (3-dimensional!) is known, minimizing the number of potential test compounds Target structure Z X N H C Y Factors Affecting The Affinity Of A Small Molecule For A Target Protein LIGAND.wat n +PROTEIN.wat n LIGAND.PROTEIN.watp+(n+m-p) wat • HYDROGEN BONDING • HYDROPHOBIC EFFECT • ELECTROSTATIC INTERACTIONS • VAN DER WAALS INTERACTIONS DIFFERENCE BETWEEN AN INHIBITOR AND DRUG Extra requirement of a drug compared to an inhibitor •Selectivity LIPINSKI’S RULE OF FIVE Poor absorption or permeation are more •Less Toxicity likely when : •Bioavailability -There are more than five H-bond donors •Slow Clearance -The mol.wt is over 500 Da •Reach The Target -The MlogP is over 4.15(or CLOG P>5) •Ease Of Synthesis -The sums of N’s and O’s is over 10 •Low Price •Slow Or No Development Of Resistance •Stability Upon Storage As Tablet Or Solution •Pharmacokinetic Parameters •No Allergies Mecanismo antibacteriano de la PZA: Pro-droga THERMODYNAMICS OF RECEPTOR-LIGAND BINDING •Proteins that interact with drugs are typically enzymes or receptors. •Drug may be classified as: substrates/inhibitors (for enzymes) agonists/antagonists (for receptors) •Ligands for receptors normally bind via a non-covalent reversible binding. •Enzyme inhibitors have a wide range of modes:non-covalent reversible,covalent reversible/irreversible or suicide inhibition. •Inhibitors are designed to bind with higher affinity: their affinities often exceed the corresponding substrate affinities by several orders of magnitude! •Agonists are analogous to enzyme substrates: part of the binding energy may be used for signal transduction, inducing a conformation or aggregation shift. •To understand ‘what forces’ are responsible for ligands binding to Receptors/Enzymes, •The observed structure of Protein is generally a consequence of the hydrophobic effect! •Proteins generally bury hydrophobic residues inside the core,while exposing hydrophilic residues to the exterior Salt-bridges inside •Ligand building clefts in proteins often expose hydrophobic residues to solvent and may contain partially desolvated hydrophilic groups that are not paired: Docking Methods • Docking of ligands to proteins is a formidable problem since it entails optimization of the 6 positional degrees of freedom. • Rigid vs Flexible • Manual Interactive Docking Automated Docking Methods • Speed vs Reliability • Basic Idea is to fill the active site of the Target protein with a set of spheres. • Match the centre of these spheres as good as possible with the atoms in the database of small molecules with known 3-D structures. • Examples: – DOCK, CAVEAT, AUTODOCK, LEGEND, ADAM, LINKOR, LUDI. GRID Based Docking Methods • Grid Based methods – GRID (Goodford, 1985, J. Med. Chem. 28:849) – GREEN (Tomioka & Itai, 1994, J. Comp. Aided. Mol. Des. 8:347) – MCSS (Mirankar & Karplus, 1991, Proteins, 11:29). • Functional groups are placed at regularly spaced (0.3-0.5A) lattice points in the active site and their interaction energies are evaluated. Folate Biosynthetic pathway DHFR CLUSTAL W (1.81) multiple sequence alignment chabaudi vinckei berghei yoelii vivax falciparum -----------------------E--KAGCFSNKTFKGLGNEGGLPWKCNSVDMKHFSSV -----------AICACCKVLNSNE--KASCFSNKTFKGLGNAGGLPWKCNSVDMKHFVSV MEDLSETFDIYAICACCKVLNDDE--KVRCFNNKTFKGIGNAGVLPWKCNLIDMKYFSSV -----------AICACCKVINNNE--KSGSFNNKTFNGLGNAGMLPWKYNLVDMNYFSSV MEDLSDVFDIYAICACCKVAPTSEGTKNEPFSPRTFRGLGNKGTLPWKCNSVDMKYFSSV -------------------------KKNEVFNNYTFRGLGNKGVLPWKCNSLDMKYFCAV * *. **.*:** * **** * :**::* :* 35 47 58 47 60 35 chabaudi vinckei berghei yoelii vivax falciparum TSYVNETNYMRLKWKRDRYMEK---------NNVKLNTDGIPSVDKLQNIVVMGKASWES TSYVNENNYIRLKWKRDKYIKE---------NNVKVNTDGIPSIDKLQNIVVMGKTSWES TSYINENNYIRLKWKRDKYMEKHNLK-----NNVELNTNIISSTNNLQNIVVMGKKSWES TSYVNENNYIRLQWKRDKYMGKNNLK-----NNAELNNGELN--NNLQNVVVMGKRNWDS TTYVDESKYEKLKWKRERYLRMEASQGGGDNTSGGDNTHGGDNADKLQNVVVMGRSSWES TTYVNESKYEKLKYKRCKYLNKET----------VDNVNDMPNSKKLQNVVVMGRTNWES *:*::*.:* :*::** :*: * .:***:****: .*:* 86 98 113 100 120 85 chabaudi vinckei berghei yoelii vivax falciparum IPSKFKPLQNRINIILSRTLKKEDLAKEYN------NVIIINSVDDLFPILKCIKYYKCF IPSKFKPLENRINIILSRTLKKENLAKEYS------NVIIIKSVDELFPILKCIKYYKCF IPKKFKPLQNRINIILSRTLKKEDIVNENN--NENNNVIIIKSVDDLFPILKCTKYYKCF IPPKFKPLQNRINIILSRTLKKEDIANEDNKNNENGTVMIIKSVDDLFPILKAIKYYKCF IPKQYKPLPNRINVVLSKTLTKEDVK---------EKVFIIDSIDDLLLLLKKLKYYKCF IPKKFKPLSNRINVILSRTLKKEDFD---------EDVYIINKVEDLIVLLGKLNYYKCF ** ::*** ****::**:**.**:. * **..:::*: :* :***** 140 152 171 160 171 136 chabaudi vinckei berghei yoelii vivax falciparum I----------------------------------------------------------IIGGASVYKEFLDRNLIKKIYFTRINNAYT-----------------------------IIGGSSVYKEFLDRNLIKKIYFTRINNSYNCDVLFPEINENLFKITSISDVYYSNNTTLD IIGGSYVYKEFLDRNLIKKIYFTRINNSYN-----------------------------IIGGAQVYRECLSRNLIKQIYFTRINGAYPCDVFFPEFDESQFRVTSVSEVYNSKGTTLD I----------------------------------------------------------* 141 182 231 190 231 137 chabaudi vinckei berghei yoelii vivax falciparum ----------------FIIYSKTKE 240 --------FLVYSKVGG 240 --------- Multiple alignment of DHFR of Plasmodium species Drug binding pocket of L. casei DHFR Antifolate drugs in the active site of DHFR L. casei to show hydrogen bonding with surrounding residues MTX TMP PYR SO3 How molecular modeling could be used in identifying new leads • These two compounds a triazinobenzimidazole & a pyridoindole were found to be active with high Ki against recombinant wild type DHFR. • Thus demonstrate use of molecular modeling in malarial drug design. Sitio Activo de la pirazinamidasa Docking P. Horikoshii – PZA en presencia de Zn Additional Drug Target: glutathione-GR Glutathione-GR Additional Drug Target: Thioredoxin reductase (TrxR) How Bioinformatics Aids in Vaccine Development / Peptide Vaccine Development Using Bionformatics Approaches Emerging and re-emerging infectious diseases threats, 1980-2001 Viral - - Bolivian hemorrhagic fever-1994,Latin America Bovine spongiform encephalopathy-1986,United Kingdom Creulzfeldt-Jackob disease(a new variant V-CID)/mad cow disease-1995-96, UK/France Dengue fever-1994-97,Africa/Asia/Latin America/USA Ebola virus-1994,Gabon;1995,Zaire;1996,United States(monkey) Hantavirus-1993,United States; 1997, Argentina HIV subtype O-1994,Africa Influenza A/Beijing/32/92, A/Wuhan/359/95, HS:N1-1993,United States; 1995,China; 1997, Hongkong Japanese Encephalitis-1995, Australia Lassa fever-1992,Nigeria Measles-1997, Brazil Monkey pox-1997,Congo Morbillivirus – 1994, Australia O’nyong-nyong fever-1996,Uganda Polio-1996,Albania Rift Valley fever-1993,Sudan Venezuelan equine encephalitis-1995-96,Venezuela/Colombia West Nile Virus-1996,Romania Yellow fever-1993,Kenya;1995,Peru Emerging and re-emerging infectious diseases threats contd., • Parasitic - African trypanosomiasis-1997,Sudan - Ancylcostoma caninum(eosinophilic enteritis)1990s,Australia - Cryptosporiadiasis-1993+,United States - Malaria-1995-97,Africa/Asia/Latin America/United states - Metorchis-1996,Canada - Microsporidiosis-Worldwide • Fungal - Coccidiodomycosis-1993,United States - Penicillium marneffi Emerging and re-emerging infectious diseases threats contd. • Bacterial – Anthrax-1993,Caribbean – Cat scratch disease/Bacillary angiomatosis(Bartonella henseiae)-1900s, USA – Chlamydia pneumoniae(Pneumonia/Coronary artery disease?)-1990s, USA(discovered 1983) – Cholera-1991,Latin America – Diphtheria-1993,Former Soviet Union – Ehrlichia chaffeensis,Human monocytic ahrlichiosis(HME)-United States – Ehrlichia phagocytophilia,Human Granulocytic ehrlichis(HGE)-United States – Escherichia coli O157-1982-1997,United States;1996,Japan – Gonorrhea(drug resistant)-1995,United States – Helicobacter pylori(ulcers/cancer_-worldwide(discovered 1983) – Leptospirosis-195,Nicaragun – Lyme disease(Borrelia burgdorferi)-1990s,United states – Meningococcal meningitis(serogroup A)-1995-1997,West Africa – Pertussis-1994,UK/Netherlands;1996,USA – Plague-1994,India – Salmonella typhimurium DT104(drug resistant)-1995,USA – Staphylococcus aureus(drug resistant)-1997,United States/Japan – Toxic strep-United States – Trench fever(Barnionella quintana)-1990s,United States – Tuberculosis(highly transmissible)-1995,United states – Vibrio cholerae 0139-1992,Southern Asia Types of Vaccines • • • • • • • Killed virus vaccines Live-attenuated vaccines Recombinant DNA vaccines Genetic vaccines Subunit vaccines Polytope/multi-epitope vaccines Synthetic peptide vaccines Systems with potential use as T-cell vaccines CD4 + T-cell vaccines Killed microbe Live attenuated microbe Synthetic peptide coupled to protein Recombinant microbial protein bearing CD4+ T-cell epitope CD8+ T-cell vaccines Live attenuated microbe Synthetic peptide delivered in liposomes or ISCOMs - Chimeric virus expressing CD4+ T-cell epitope Chimeric virus expressing CD8+ T-cell epitope Chimeric Ig Self-molecule expressing CD8+ T-cell epitope Chimeric-peptide-MHC class II complex Chimeric peptide-MHC Class I complex Receptor-linked peptide Naked DNA expressing CD4+ T-cell epitope Naked DNA expressing CD8+ T-cell epitope Abbreviations: Ig, Immunoglobulin, ISCOM, immune-stimulating complex; MHC,Major histocompability complex. Why Synthetic Peptide Vaccines? Chemically well defined, selective and safe. Stable at ambient temperature. No cold chain requirement hence cost effective in tropical countries. Simple and standardised production facility. What Are Epitopes? Antigenic determinants or Epitopes are the portions of the antigen molecules which are responsible for specificity of the antigens in antigen-antibody (Ag-Ab) reactions and that combine with the antigen binding site of Ab, to which they are complementary. Epitopes could be contiguous (when Ab binds to a contiguous sequence of amino acids) non-contiguous (when Ab binds to non-contiguous residues, brought together by folding). Sequential epitopes are contiguous epitopes. Conformational epitopes are noncontiguous antigenic determinants. Epitopes … B-cell epitopes Th-cell epitopes Properties of Amino Acids: predictors for Epitopes Sequential epitope prediction methods Theoretical methods are based on properties of amino acids and their propensity scales. Hopp & Woods, 1981. Parker et al., 1986 Kolaskar & Tongaonkar, 1990. The accuracy of prediction: 50-75%. Conformational epitope prediction method Kolaskar & Kulkarni-Kale, 1999. Identified antigens must be checked for strain varying polymorphisms, these polymorphism must be represented in a anti-blood stage vaccine Protective epitope Variants in strains A B C Candidate protein X D Antigenic determinants of Egp of JEV Kolaskar & Tongaonkar approach Peptide vaccines to be launched in near future • • • • • • • Foot & Mouth Disease Virus (FMDV) Human Immuno Deficiency Virus (HIV) Metastatic Breast Cancer Pancreatic Cancer Melanoma Malaria * T.solium cysticercosis * Various transformations on side-chain orientation in a model tetrapeptide Reverse Vaccinology • Advantages – – – – – Fast access to virtually every antigen Non-cultivable can be approached Non abundant antigens can be identified Antigens not expressed in vitro can be identified. Non-structural proteins can be used • Disadvantages – Non proteinous antigens like polysaccharides, glycolipids cannot be used. Rappuoli 2001 Curr. Opin. Microbiol. Rappuoli 2001 Curr. Opin. Microbiol. Vaccine development In Post-genomic era: Reverse Vaccinology Approach. Genome Sequence Proteomics Technologies In silico analysis IVET, STM, DNA microarrays High throughput Cloning and expression In vitro and in vivo assays for Vaccine candidate identification Global genomic approach to identify new vaccine candidates In Silico Analysis Peptide Multitope vaccines VACCINOME Candidate Epitope DB Epitope prediction Disease related protein DB Gene/Protein Sequence Database Synthetic Peptide Vaccine Design and Development of Synthetic Peptide vaccine against Japanese encephalitis virus Egp of JEV as an Antigen Is a major structural antigen. Responsible for viral haemagglutination. Elicits neutralising antibodies. ~ 500 amino acids long. Structure of extra-cellular domain (399) was predicted using knowledge-based homology modeling approach. Model Refinement PARAMETERS USED • force field: • Dielectric const: • Optimisation: AMBER all atom Distance dependent Steepest Descents & Conjugate Gradients. • rms derivative 0.1 kcal/mol/A for SD • rms derivative 0.001 kcal/mol/A for CG • Biosym from InsightII, MSI and modules therein Model For Solvated Protein Egp of JEV molecule was soaked in the water layer of 10A. 4867 water molecules were added. The system size was increased to 20,648 atoms from 6047. Model Evaluation II: Ramachandran Plot An Algorithm to Identify Conformational Epitopes Calculate the percent accessible surface area (ASA) of the amino acid residues. If ASA 30%, then residue was termed as accessible residues. A contiguous stretch of more than three accessible residues was termed as the antigenic determinant. …Cont. A determinant is extended to N- and Cterminals, only if, accessible amino acid(s) are present after an inaccessible amino acid residue. A list of sequential antigenic determinants was prepared. Peptide Modeling Initial random conformation Force field: Amber Distance dependent dielectric constant 4rij Geometry optimization: Steepest descents & Conjugate gradients Molecular dynamics at 400 K for 1ns Peptides are: SENHGNYSAQVGASQ NHGNYSAQVGASQ YSAQVGASQ YSAQVGASQAAKFT NHGNYSAQVGASQAAKFT SENHGNYSAQVGASQAAKFT 149 168 Prediction of conformations of the antigenic peptides Lowest energy Allowed conformations were obtained using multiple MD simulations: – Initial conformation: random, allowed – Amber force field with distance dependent dielectric constant of 4*rij – Geometry optimization using Steepest descents & Conjugate gradient – 10 cycles of molecular dynamics at 400 K; each of 1ns duration, with an equilibration for 500 ps – Conformations captured at 10ps intervals, followed by energy minimization of each – Analysis of resulting conformations to identify the lowest energy, geometrically and stereochemically allowed conformations MD simulations of following peptides were carried out B Cell Epitopes: SENHGNYSAQVGASQ NHGNYSAQVGASQ YSAQVGASQ YSAQVGASQAAKFT NHGNYSAQVGASQAAKFT 149 T-helper Cell Epitope: 436 445 SIGKAVHQVF 168 SENHGNYSAQVGASQAAKFT Chimeric B+Th Cell Epitope With Spacer: SENHGNYSAQVGASQAAKFTSIGKAVHQVF Structural comparison of Egps of Nakayama and Sri Lanka strains of JEV. Single amino acid differences are highlighted. Ts18 epitope mapping 1.6 1.6 1.2 1.2 1.2 0.8 0.4 0.8 0.0 0.0 1 3 5 7 9 11 13 15 17 19 0.8 0.4 0.4 0.0 1 3 5 7 9 11 13 15 17 1 19 1.6 1.6 1.2 1.2 1.2 0.8 0.8 0.4 0.4 0.0 0.0 1 3 5 7 9 11 13 15 17 19 A650 1.6 A650 A650 A650 1.6 A650 A650 13-mers window skipping 3 aminoacids 3 5 7 9 11 13 11 13 15 15 17 0.8 0.4 0.0 1 3 5 7 9 11 13 15 17 19 1 3 5 7 9 17 19 19 Ts18 MHC II epitope profiles for different alleles Ts18 MHC I and MHC II consensus profile 45 40 35 30 25 20 15 10 5 0 1 5 9 13 17 21 25 29 33 37 41 45 49 53 57 61 65 69 73 Ts18 modeled 3D structure