Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Nutriepigenomics wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Genetic testing wikipedia , lookup
Pathogenomics wikipedia , lookup
Gene expression profiling wikipedia , lookup
Minimal genome wikipedia , lookup
Gene expression programming wikipedia , lookup
Genetic drift wikipedia , lookup
Dominance (genetics) wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Genomic imprinting wikipedia , lookup
Genetic engineering wikipedia , lookup
Medical genetics wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Genome evolution wikipedia , lookup
Polymorphism (biology) wikipedia , lookup
History of genetic engineering wikipedia , lookup
Biology and consumer behaviour wikipedia , lookup
Public health genomics wikipedia , lookup
Human genetic variation wikipedia , lookup
Genome (book) wikipedia , lookup
Population genetics wikipedia , lookup
Designer baby wikipedia , lookup
Microevolution wikipedia , lookup
Behavioural genetics wikipedia , lookup
Chapter 22 - Quantitative genetics:
•
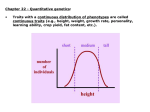
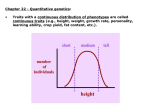
Traits with a continuous distribution of phenotypes are called
continuous traits (e.g., height, weight, growth rate, personality,
learning ability, crop yield, fat content, etc.).
Continuous traits arise from effects of:
•
Multiple loci (effects of many genes)
•
Pleiotropy (one gene has many effects)
•
Epistasis (gene interaction effects)
•
Variable expressivity and penetrance
•
Environment (produces a range of phenotypes)
Continuous traits:
•
Francis Galton and Karl Pearson (late 1800s):
Recognized that continuous traits are statistically correlated
between parents and offspring, but could not determine how
transmission occurs.
•
Wilhelm Johannsen (1903):
Demonstrated that bean seed weight is partly heritable and partly
environmental.
•
Sir Ronald Fisher (1890-1962):
First to demonstrate mathematically that Mendelian models of allele
segregation apply to multiple genetic loci.
Types of questions studied in quantitative genetics:
•
How do genetics and the environment affect a trait?
•
Which and how many genes produce a set of phenotypes for a trait;
where in the genome are they located?
•
Do some genes play a major role, whereas other genes modify or
play a small role?
•
Do alleles interact to produce additive or epistatic effects?
•
How does selection affect the trait; does it affect other traits that
may be linked?
•
If so what types of mating and selection produce desired
phenotypes?
Types of data collected and analyses to consider:
•
Sample size/randomization
•
Type of distribution (e.g., normal distribution)
•
Mean, variance, and standard deviation
•
Correlation/regression
•
Analysis of variance (ANOVA)
Lognormal
Normal
Exponential
Quantitative trait loci (QTLs):
QTLs =specific genomic segments correlated with continuous
phenotypic trait variation.
Perform Genome Wide Association Study (GWAS)
1.
Cross inbred lines with different phenotypes (homozygotes for
different alleles at most loci) to produce heterozygotes.
2.
Self F1 or back cross to parental lines to increase phenotypic
variation and segregation of traits.
3.
Analyze F2 with physical markers (microsatellites) that correlate
with phenotypic variation.
4.
Create a linkage map.
5.
Calculate components of phenotypic variance (VP) due to genetic
effects (VG) and components due to environment effects (VE).
VP = VG + VE + 2COVG,E + VG x E
Components of genetic variance:
1.
Additive genetic variance (VA): effects of alleles at two or more loci
contribute to phenotype. F1 will will appear intermediate to the
parental phenotypes for repeated test crosses.
2.
Dominance variance (VD): effects of alleles are not strictly additive;
must consider how alleles interact in the heterozygote. F1 will
resemble one of the parental phenotypes.
3.
Interaction variance (VI): accounts for epistatic interactions
between two or more loci. F1 phenotype is unpredictable.
VG = VA + V D + V I
VP = (VA + VD + VI) + VE + 2COVG,E + VG x E
Calculate Lod Score (Logarithm of the Odds):
1.
Lod = log of the ratio odds that two loci (or a locus and a trait) are
linked with a recombination factor (q) greater than 0 and less than
0.5.
2.
z = log10 {Prob(data|q)/Prob(data|0.5)}
3.
Lod score of +3.00 (odds of 1000:1) or greater is regarded as
acceptable evidence for linkage.
Lod 3 = 1000:1
Lod 2 = 100:1
Lod 1 = 10:1
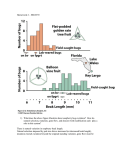
Figure 1. Graph of multipoint lod scores assuming heterogenity
The peak multipoint lod score of 3.85 is located between DXS1200 and DXS297.
Nature Genetics 20, 175 - 179 (1998)
Evidence for a prostate cancer susceptibility locus on the X chromosome.
Jianfeng Xu et al.
Timmerman-Vaughan et al. 2004 - Linkage mapping of QTLs for seed yield, yield components
and developmental traits in pea (Pisum sativum L.)
Chromosome map of human QTLs for plasma concentrations of HDL-C, LDL-C and triglyceride
levels. 2007. The Jackson Laboratory
Broad- and Narrow-Sense Heritability:
1.
Broad-sense heritability
= hB2 = VG/VP
2.
Narrow-sense heritability
= hN2 = VA/VP
*Broad-sense heritability measures proportion of phenotypic variance
among individuals in a population that results from genetic
differences.
*Narrow-sense heritability measures proportion of phenotypic variance
that results from additive genetic variance only.
*Narrow sense heritability is what can be used to predict resemblance
between offspring and parents, between you and your parents.
*Heritability is a measure of variance and is only meaningful for
characteristics of a population (not the individual).
Example showing how to calculate narrow-sense heritability using
parent-offspring regression:
Example showing response to selection in artificial experiment:
h2 = R/S R = h2S
Genome Scan
•Search for islands of genetic differentiation in otherwise
undifferentiated genetic background.
•Method of searching for genes for functionally important traits.
•Does not require crossing experiment, but rather perform genomic scan
(e.g., next-generation sequencing) for two populations that differ in a
single environmental variable subject to strong selection.
•Works best for two populations that are in migration-selection balance
equilibrium experiencing strong divergent selection and high gene flow.
•Utilize measures such as Sewall Wright’s Fst
•Examines linked patterns of statistically correlated divergence in
different genes, which may result from correlated selection and/or
divergence hitchhiking through depressed recombination.
Alternate interpretations of outliers.
Via S Phil. Trans. R. Soc. B 2012;367:451-460
©2012 by The Royal Society
Threespine Stickleback
Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson
EA, et al. 2010 Population Genomics of Parallel
Adaptation in Threespine Stickleback using Sequenced
RAD Tags. PLoS Genet 6(2): e1000862.
doi:10.1371/journal.pgen.1000862
Multiple genes in same region evolve as a cassette
(recombination is suppressed)
©2012 by The Royal Society
Via S Phil. Trans. R. Soc. B 2012;367:451-460
Probable DH regions in threespine stickleback.