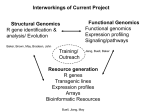
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Epigenetics of human development wikipedia , lookup
Segmental Duplication on the Human Y Chromosome wikipedia , lookup
Gene expression programming wikipedia , lookup
Gene nomenclature wikipedia , lookup
History of genetic engineering wikipedia , lookup
Gene therapy of the human retina wikipedia , lookup
Adeno-associated virus wikipedia , lookup
Minimal genome wikipedia , lookup
Gene desert wikipedia , lookup
Genome (book) wikipedia , lookup
DNA barcoding wikipedia , lookup
Gene expression profiling wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Koinophilia wikipedia , lookup
Human genome wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Non-coding DNA wikipedia , lookup
Helitron (biology) wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Genomic library wikipedia , lookup
Microevolution wikipedia , lookup
Designer baby wikipedia , lookup
Genome evolution wikipedia , lookup
Metagenomics wikipedia , lookup
Comparative Genomics Bioinformatic Tools for Comparative Genomics of Vectors Overview Comparing Genomes Homologies and Families Sequence Alignments Bioinformatic Tools for Comparative Genomics of Vectors Comparative Genomics Allows us to achieve a greater understanding of vertebrate evolution Tells us what is common and what is unique between different species at the genome level The function of human genes and other regions may be revealed by studying their counterparts in lower organisms Helps identify both coding and non-coding genes and regulatory elements Bioinformatic Tools for Comparative Genomics of Vectors Sequence Conservation Over Time Bioinformatic Tools for Comparative Genomics of Vectors Non Coding Regions Large stretches of non-coding regions in vertebrates Regulatory regions of: Developmental genes Transcription factors miRNA Bioinformatic Tools for Comparative Genomics of Vectors Kikuta et al., Genome Research, May 2007 Methods of Alignment- Ensembl BLASTZ-net (comparison on nucleotide level) is used for species that are evolutionary close, e.g. human – mouse Translated BLAT (comparison on amino acid level) is used for evolutionary more distant species, e.g. human – zebrafish PECAN global alignment used for multispecies alignments Bioinformatic Tools for Comparative Genomics of Vectors Why Compare Genomes? We can better understand evolution/ speciation We can find important, functional regions of the sequence (codons, promoters, regulatory regions) It can help us locate genes in other species that are missing or not welldefined (also through comparison and alignments). Quality control! Bioinformatic Tools for Comparative Genomics of Vectors Evolution at the DNA Level Deletion Mutation …ACTGACATGTACCA… Sequence edits …AC----CATGCACCA… Rearrangements Inversion Translocation Duplication Bioinformatic Tools for Comparative Genomics of Vectors Comparing Genomes • • • Mammals have roughly 3 billion base pairs in their genomes Over 98% human genes are shared with primates, with more than 9598% similarity between genes. Even the fruit fly shares 60% of its genes with humans! (March 2000) • Compare human & Mouse • • 40% of human genome align with mouse 24% of human genome missing in mouse (also mouse-specific sequences) Bioinformatic Tools for Comparative Genomics of Vectors Improving Gene Quality Comparative genomics predicts one long transcript. Bioinformatic Tools for Comparative Genomics of Vectors Pseudogene recovery chr 3 human mouse rat dog cow chr X We find 67 confident cases where a human protein is closer to the ancestor than any extant species in the alignment Bioinformatic Tools for Comparative Genomics of Vectors How Does Ensembl Predict Homology? • Uses all the species • Prediction pipeline: Begins with BLAST and sequence clustering • Compares gene relationships to species relationships Bioinformatic Tools for Comparative Genomics of Vectors BSR: Blast Score Ratio. When 2 proteins P1 and P2 are compared, BSR=scoreP1P2/max(selfscoreP1 or self-scoreP2). The default threshold used in the initial clustering step is 0.33. Orthologue / Paralogue Prediction Algorithm (1) Load the longest translation of each gene from all species used in Ensembl. (2) Run WUBLASTp+SW of every gene against every other (both self and non-self species) in a genome-wide manner. (3) Build a graph of gene relations based on Best Reciprocal Hits (BRH) and Blast Score Ratio (BSR) values. (4) Extract the connected components (=single linkage clusters), each cluster representing a gene family. (5) For each cluster, build a multiple alignment based on the protein sequences using MUSCLE. (6) For each aligned cluster, build a phylogenetic tree using PHYML. An unrooted tree is obtained at this stage. (7) Reconcile each gene tree with the species tree to call duplication event on internal nodes and root the tree (TreeBeSt). (8) From each gene tree, infer gene pairwise relations of orthology and paralogy types. Bioinformatic Tools for Comparative Genomics of Vectors Species Tree Anopheles gambiae Aedes aegypti Drosophila melanogaster Dasypus novemcinctus Loxodonta africana Echinops telfairi Tupaia belangeri Homo sapiens Pan troglodytes Macaca mulatta Otolemur garnettii Mus musculus Rattus norvegicus Spermophilus tridecemlineatus Cavia porcellus Oryctolagus cuniculus Erinaceus europaeus Myotis lucifugus Canis familiaris Felis catus Bos taurus Monodelphis domestica Ornithorhynchus anatinus Gallus gallus Xenopus tropicalis Gasterosteus aculeatus Oryzias latipes Takifugu rubripes Tetraodon nigroviridis Danio rerio Ciona intestinalis Ciona savignyi Caenorhabditis elegans Saccharomyces cerevisiae Bioinformatic Tools for Comparative Genomics of Vectors Species and Gene Trees Phylogenetic Tree Reconciliation: the Species/Gene Tree Problem Dufayard et al. ERCIM News No. 43 October 2000 Bioinformatic Tools for Comparative Genomics of Vectors Genes/Species Tree reconciliation: TreeBeST Bioinformatic Tools for Comparative Genomics of Vectors Reconciliation M M Duplication node Speciation node R R H species tree H M M R’ H H R H’ unrooted gene tree M’ R Viewing Trees in Ensembl GeneView page GeneTreeView Bioinformatic Tools for Comparative Genomics of Vectors Types of Homologues Orthologs : any gene pairwise relation where the ancestor node is a speciation event Paralogs : any gene pairwise relation where the ancestor node is a duplication event Bioinformatic Tools for Comparative Genomics of Vectors Orthologue and Paralogue Types ortholog_one2one ortholog_one2many ortholog_many2many apparent_ortholog_one2one within_species_paralog between_species_paralog Bioinformatic Tools for Comparative Genomics of Vectors Ortholog and Paralog types Ortholog and Paralog types Bioinformatic Tools for Comparative Genomics of Vectors Orthologues on GeneView What is ‘1 to 1’? What is ‘1 to many’? Bioinformatic Tools for Comparative Genomics of Vectors Protein Families How: Cluster proteins for every isoform (transcript) in every species. Why: Predict a function for ‘novel’ genes/proteins Understand gene relationships Bioinformatic Tools for Comparative Genomics of Vectors Protein Dataset More than 1,800,000 proteins clustered: All Ensembl protein predictions from all species supported 895,070 protein predictions All metazoan (animal) proteins in UniProt: 96,030 UniProtKB/Swiss-Prot 892,0208 UniProtKB/TrEMBL Bioinformatic Tools for Comparative Genomics of Vectors Clustering Strategy BLASTP all-versus-all comparison Markov clustering For each cluster: Calculation of multiple sequence alignments with ClustalW Assignment of a consensus description Bioinformatic Tools for Comparative Genomics of Vectors Where are Families shown? ProtView Link to FamilyView Bioinformatic Tools for Comparative Genomics of Vectors Where are Families shown? FamilyView JalView multiple alignments Ensembl family members within human Ensembl family members in other species Bioinformatic Tools for Comparative Genomics of Vectors Comparing Genomes Homologies and Families Sequence alignments Bioinformatic Tools for Comparative Genomics of Vectors Aligning Whole Genomes- Why? • To identify homologous regions • To spot trouble gene predictions • Conserved regions could be functional • To define syntenic regions (long regions of DNA sequences where order and orientation is highly conserved) Bioinformatic Tools for Comparative Genomics of Vectors Aligning large genomic sequences Should find all highly similar regions between two sequences Should allow for segments without similarity, rearrangements etc. Issues Heavy process Scalability, as more and more genomes are sequenced Time constraint Bioinformatic Tools for Comparative Genomics of Vectors Whole Genome Multiple Alignments Enredo Defines orthology map (co-linear regions) Supports segmental duplications Pecan Consistency based multiple aligner Optimized to cope with long DNA sequences Ortheus Ancestral sequences reconstructor Inferring the history of insertion and deletions In ContigView... Bioinformatic Tools for Comparative Genomics of Vectors Multiple Alignments using PECAN Currently 2 sets: 10 amniota vertebrates: 7 eutherian mammals: To come… the fish! Bioinformatic Tools for Comparative Genomics of Vectors Alignment Strategy Use all coding exons Get sets of best reciprocal hits Create orthology maps Build multiple global alignments Bioinformatic Tools for Comparative Genomics of Vectors View Alignments: ContigView In the Detailed View Panel: Bioinformatic Tools for Comparative Genomics of Vectors View Conservation: ContigView Click on a Pink Bar for AlignSliceView… export alignments Bioinformatic Tools for Comparative Genomics of Vectors AlignSliceView Bioinformatic Tools for Comparative Genomics of Vectors GeneSeqalignView Bioinformatic Tools for Comparative Genomics of Vectors GeneSeqalignView Bioinformatic Tools for Comparative Genomics of Vectors MultiContigView Comparison of chromosomes in multiple species. (Links from SyntenyView, ContigView, CytoView) Bioinformatic Tools for Comparative Genomics of Vectors Export Alignments in BioMart Choose ‘Compara pairwise alignments’ Bioinformatic Tools for Comparative Genomics of Vectors Syntenic Regions Genome alignments are compiled into larger syntenic regions Alignments are clustered together when the relative distance between them is less than 100 kb and order and orientation are consistent Any clusters less than 100 kb are discarded Bioinformatic Tools for Comparative Genomics of Vectors Enredo Anchors 500.000 anchors for mammals --more than 1 anchor per 10Kb Supports segmental duplications!! Covers 90% of the human protein coding genes (Hsap-Mmus-Rnor-Cfam-Btau) Bioinformatic Tools for Comparative Genomics of Vectors SyntenyView Human chromosome Orthologues Mouse chromosomes Mouse chromosomes Bioinformatic Tools for Comparative Genomics of Vectors CytoView Syntenic blocks Bioinformatic Tools for Comparative Genomics of Vectors Summary View Homology in pages such as GeneView, ProtView, SyntenyView, GeneTreeView, or BioMart View Protein Family information in FamilyView View Alignments in ContigView, GeneSeqAlign View, through BioMart Bioinformatic Tools for Comparative Genomics of Vectors