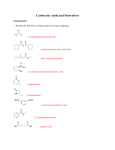
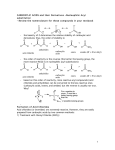
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Bottromycin wikipedia , lookup
Woodward–Hoffmann rules wikipedia , lookup
George S. Hammond wikipedia , lookup
Asymmetric induction wikipedia , lookup
Physical organic chemistry wikipedia , lookup
Ring-closing metathesis wikipedia , lookup
Diels–Alder reaction wikipedia , lookup
Baylis–Hillman reaction wikipedia , lookup
Ene reaction wikipedia , lookup
Wolff rearrangement wikipedia , lookup
Tiffeneau–Demjanov rearrangement wikipedia , lookup
Hofmann–Löffler reaction wikipedia , lookup
Hydroformylation wikipedia , lookup
Wolff–Kishner reduction wikipedia , lookup
Petasis reaction wikipedia , lookup
The Institute of Paper Chemistry Appleton, Wisconsin Doctor's Dissertation Anhydride Derivatives of Trimellitic Anhydride Richard G. Barker June, 1963 ANHYDRIDE DERIVATIVES OF TRIMELLITIC ANHYDRIDE A thesis submitted by Richard G. Barker B.A. 1958, Hamilton College M.S. 1960, Lawrence College in partial fulfillment of the requirements of The Institute of Paper Chemistry for the degree of Doctor of Philosophy from Lawrence College Appleton, Wisconsin June, 1963 TABLE OF CONTENTS Page GENERAL SUMMARY 1 INTRODUCTION 3 Trimellitic Anhydride 3 Unsymmetrical Anhydrides 5 Preparation of Unsymmetrical Anhydrides 6 Disproportionation of Unsymmetrical Anhydrides 8 Hydrolysis of Unsymmetrical Anhydrides 11 Reactions of Unsymmetrical Anhydrides 12 Previous Investigations on the Esterification of Unsymmetrical Anhydrides 12 The Mechanism of Acylation by Unsymmetrical Anhydrides 13 Trifluoroacetic Anhydride as an Esterification Promoter 16 PRESENTATION AND APPROACH TO THE PROBLEM 22 PROCEDURES AND RESULTS 24 The Trimellitic Anhydride-Trifluoroacetic Anhydride Reaction System 24 Preparation of the Unsymmetrical Anhydride of TMA and TFAA 24 Purification and Analysis of the Unsymmetrical Anhydride of TMA and TFAA 25 Disproportionation of the Unsymmetrical Anhydride of TMA and TFAA 27 Hydrolysis of the Unsymmetrical Anhydride of TMA and TFAA 28 Esterification of the Unsymmetrical Anhydride of TMA and TFAA 29 The Unsymmetrical Anhydride of Trimellitic and Acetic Anhydrides 31 Preparation of the Unsymmetrical Anhydride 31 Purification and Analysis of the Unsymmetrical Anhydride 32 Reaction of the Unsymmetrical Anhydride with Methanol 35 iii Page The Acid Chloride of Trimellitic Anhydride 36 Reaction of TMA with Thionyl Chloride 36 Formation of the Trianhydride 37 Formation of the Acid Chloride of Trimellitic Anhydride 37 Purification and Analysis of the Acid Chloride 38 Reaction of the Acid Chloride and Alcohols 38 Preliminary Esterifications 39 Quantitative Study 41 Analysis of 4-Carbomethoxy Phthalic Anhydride 41 Esterification of the Trianhydride with Alcohols 42 Preliminary Reactions 42 Quantitative Study 45 DISCUSSION OF RESULTS AND CONCLUSIONS The Trimellitic Anhydride-Trifluoroacetic Anhydride Reaction System 46 46 The Unsymmetrical Anhydride of TMA and TFAA 46 Esterification of the TMA-TFAA System 50 The Unsymmetrical Anhydride of Trimellitic and Acetic Anhydrides 57 The Acid Chloride of Trimellitic Anhydride 59 .Bis(Trimellitic Anhydride) Anhydride 61 Summary of Results 62 ACKNOWLEDGMENTS 64 LITERATURE CITED 65 APPENDIX I. 70 APPENDIX II. APPENDIX III. DETERMINATION OF ANHYDRIDE EQUIVALENT DETERMINATION OF MOLECULAR WEIGHT FLUORINE ANALYSIS 72 73 iv Page APPENDIX IV. ESTERIFICATION REACTIONS OF ALCOHOLS WITH THE TRIMELLITIC ANHYDRIDE-TRIFLUOROACETIC ANHYDRIDE SYSTEM 75 APPENDIX V. PREPARATION OF 4-CARBOMETHOXY PHTHALIC ANHYDRIDE FROM TMA AND DIAZOMETHANE 93 APPENDIX VI. ANHYDRIDE 95 METHYLATION OF THE ACID CHLORIDE OF TRIMELLITIC 1 GENERAL SUMMARY Anhydride derivatives of trimellitic anhydride, which possesses both a free carboxyl group and a cyclic anhydride group, were prepared. Substitution of the carboxyl group with a more reactive reagent could lead to specific esterification intermediates which would be of interest for the preparation of the 4-carboalkoxy phthalic anhydride series, simple and mixed diesters, and cross-linked esters. The unsymmetrical anhydride with trifluoroacetic acid, a dianhydride, was prepared and characterized. This highly reactive compound readily disproportion- ates into its component simple anhydrides. Esterification reactions of various alcohols were conducted with the solid unsymmetrical anhydride, and with solutions of this anhydride in nonpolar solvents, acetone, and trifluoroacetic acid. The reactions in acetone were strongly influenced by the position of the equilibrium of the disproportionation reaction. The other reactions all appeared to follow the S 2 mechanism, rather than the ionization mechanism, resulting in the formation of trifluoroacetate esters and trimellitic anhydride. unsymmetrical anhydride with an excess of Reaction of the alcohol led to the opening of the cyclic anhydride ring, resulting in monotrimellitate esters. The unsymmetrical anhydride with acetic acid, a dianhydride, was also prepared and characterized. The methyl ester anhydride was not isolated when this unsymmetrical anhydride reacted with methanol. The monocarboxylic acid chloride of trimellitic anhydride was prepared by reaction with thionyl chloride, purified, and characterized. The reaction of the acyl chloride group with methanol was shown to be fairly specific and led to the isolation of 4-carbomethoxy phthalic anhydride. The simple anhydride of trimellitic anhydride, which is actually a trianhydride with two cyclic and one linear anhydride groups, was obtained from several -2- different reactions, purified, and characterized. The reaction of this trianhy- dride with methanol led to the predicted isolation of 4-carbomethoxy phthalic anhydride and trimellitic anhydride, but the reaction is not as specific as in the case of the reaction of the acid chloride. Both the acid chloride of trimellitic anhydride and its anhydride would be of interest in further study of the esterification intermediates. The acid chloride seems to be the most promising agent for specific esterification. -3INTRODUCTION TRIMELLITIC ANHYDRIDE Trimellitic-anhydride (the 1,2-anhydride of 1,2,4-tricarboxy benzene), hereafter referred to as TMA, contains both an aromatic acid group and a cyclic anhydride group. TMA should be interesting as an intermediate in the preparation of 0 H00C ~--C HOOC I 0/ J-C/O Trimellitic Anhydride other compounds, such as polyesters and mixed esters, but its derivatives have not been investigated very thoroughly. Derivatives of TMA which have been pre- pared are described below. Since the three carboxyl groups are not symmetrical, it is easily seen that there are three possible monoesters, one ester anhydride, three diesters, and one triester for the case of esterification with a given monohydroxy alcohol. the possible methyl esters have been prepared by various methods. All of A thorough study of the methylation of trimellitic acid was conducted by Wegscheider, et al. (1). The 1- and 2-methyl esters were prepared by boiling the acid with methanol, by heating the acid with methanol and a mineral acid, by partially hydrolyzing the trimethyl and the 1,2-dimethyl esters, and by treating methyl iodide with the appropriate silver salts. The 4-methyl ester was prepared by heating either the acid or the anhydride with methanol and by hydrolyzing the trimethyl ester. -4The ester anhydride, 4-carbomethoxy phthalic anhydride, was prepared (1) by distilling under reduced pressure the sirups obtained in the course of the different esterification reactions. Duff, et al. (2) obtained the anhydride methyl ester by treating TMA with ethereal diazomethane. Wegscheider, et al. (1) obtained 1,2-dimethyl trimellitate by partial hydrolysis of the triester, by reaction of the disilver salt with methyl iodide, and by esterification of a monoester in the presence of hydrochloric acid. along with all of the monoesters, is crystalline. This diester, The remaining diesters and the triester are sirups. The 1,4- and 2,4-diesters have been prepared by reaction of methanol with either the acid or the anhydride. Trimethyl trimellitate has been obtained by reacting methanol with the acid or anhydride in the presence of sulfuric acid (1, 3) and by reaction of methyl iodide with the trisilver salt (1). The triester has also been isolated after reaction of the acid with ethereal diazomethane (4-6). Additional esters of TMA and monohydroxy alcohols have been but briefly investigated. Triethyl trimellitate has been mentioned (7, 8). Other esters which have been obtained in yields up to 89% by reacting TMA with the alcohol in the presence of a sulfuric acid catalyst, are the tri-n-butyl, tri-n-hexyl, trioctyl, triisoctyl, and triisodecyl (9). During the course of a second year study (10), a limited investigation was made of various catalysts and their utilization in the formation of trimellitic esters of cellulose. A reaction of TMA and cellulose carried out in trifluoro- acetic anhydride led to an ester of total carboxyl content 17.6% and free carboxyl content 12.6%. An infrared spectrum of the product suggested the possibility that the ester contained unreacted anhydride groups, which would indicate that the free -5carboxyl group of TMA had been esterified with cellulose. a mixed cellulose acetate trimellitate ester. Malm (11) has mentioned Condensation polymers of TMA and glycols, epoxide resins, and alkyd resins formed by reaction with soybean oil, tall oil, and linseed oil have also been mentioned (8). Bis (trimellitic anhydride) anhydride, which is the simple anhydride of TMA, has been prepared by refluxing a solution of TMA and acetic anhydride, followed by CXO'OC 0 -c/\0 Bis (Trimellitic Anhydride) Anhydride distillation to remove the acetic acid formed and excess acetic anhydride (12). This trianhydride could be an important esterification intermediate as it can be reacted with alcohols with the accompanying formation of less water than that obtained when TMA is esterified. UNSYMMETRICAL ANHYDRIDES Anhydrides are compounds which contain the elements of two carboxyl groups less a mole of water, as in: is a simple anhydride. RCOOCOR'. If R and R' are different, the anhydride is termed a mixed or unsymmetrical anhydride. by a variety of procedures. into simple anhydrides. If R and R' are the same, the compound Unsymmetrical anhydrides have been prepared They are very reactive and disproportionate readily The reactions of various unsymmetrical anhydrides with alcohols and basic compounds have been studied to a certain extent. -6PREPARATION OF UNSYMMETRICAL ANHYDRIDES Unsymmetrical anhydrides have been known for over a century. and Chiozza (14) were the earliest investigators. Gerhardt (13) They prepared several unsym- metrical anhydrides by reacting the chloride of one acid with the alkali salt of a second acid: R COC1 + R COOK R COOCOR + KC1 (1) The silver salt of an acid was also used in an early reaction with an acid chloride (15). The triethylamine salt has been employed similarly (16). Ferris and Emmons prepared several unsymmetrical anhydrides, including benzoyl trifluoroacetate, by the reaction of silver trifluoroacetate with the appropriate acid chloride (17). A second widely used method of preparation of unsymmetrical anhydrides is the reaction of an anhydride with another acid, resulting in the equilibrium: R1 COOH + (R2C0) 2 0 ' - R1 0COCOR2 + R2 COOH (2) Autenrieth (18) and Askenasy and Meyer (19) employed this reaction to prepare various mixed acetic anhydrides. Bourne, et. aL (20) prepared various unsymmetri- cal anhydrides, including benzoyl trifluoroacetate, by the reaction of trifluoroacetic anhydride with another acid. Refluxing an acid with acetic anhydride might also result in the preparation of the simple anhydride of the other acid. Examples are the preparation of the anhydrides of isophthalic and terephthalic acids (21) and various aromatic polyanhydrides (22). The formation of a cyclic anhydride from a dicarboxylic acid by reaction with acetic anhydride probably also proceeds through the unsymmetrical anhydride with acetic anhydride (23): -7CH2COOH | CH2 COOH CH2COOH [ d CH2 CO-OCOCH3 + CH2COOH | CH2 CO-OCOCIH3 (CH3CO)20 CH2-COI | C 2-CO + CH3COOH (3) () CHC C .HI... H-OCOCHCCOOH CH2CO Unsymmetrical anhydrides have also been prepared by heating fatty acid chlorides with excess acetic anhydride (24) and by treating an acid chloride with an acid in the presence of pyridine (25). Triethylamine and N-ethylpiperidine have been substituted for pyridine in the latter case (26). Ketene has been widely employed in the preparation of unsymmetrical acetic anhydrides: RCOOH + CH2 =C=O - RCO-O-COCH3 Early investigations were conducted by Hurd, et al. (27). (4) More recently (28), thirty aliphatic and aromatic acids were acetylated with ketene in ether resulting in unsymmetrical anhydrides. Similar reactions resulting in unsymmetrical anhy- drides were carried out with carbon suboxide by Diels and Lalin (29) and with diphenylketene by Staudinger (30). Several other procedures have been suggested but have received only minor attention in the literature. Oxalyl chloride has been employed to prepare un- symmetrical anhydrides of some nitrobenzoic acids (31). Cattelain (32) prepared P-benzoylacrylic acid mixed anhydrides from y-phenylallylacetic acid via an intermediate P-iodo-y-lactone. Mixed anhydrides of monobasic carboxylic acids have also been prepared by heating alkaline salts of acyl sulfuric acids in the presence of an alkali salt of a carboxylic acid and the free acid (33). -8- Carboxylic acid chlorides have also been considered as unsymmetrical anhydrides. The diacid chlorides of terephthalic and isophthalic acids have been prepared by reaction with either thionyl chloride or phosphorus pentachloride (34). Refluxing phthalic acid with thionyl chloride results in the closing of the ring and formation of phthalic anhydride (35). The dichloride of phthalic acid has been prepared, however, by reacting phthalic anhydride with phosphorus pentachloride (36) or by treating phthalic thioanhydride with dry chlorine (37). There was no reaction when trimesic acid (benzene-l,3,5-tricarboxylic acid) was heated with thionyl chloride, but phosphorus pentachloride led to trimesoyl trichloride (38). When methyl acid phthalate was refluxed for one hour in thionyl chloride, the monochloride was obtained: COOCH3 OOCH3 + SOC12 (5) -COOH -ecOC However, with longer reflux, the cyclic ring closed leading to phthalic anhydride (39). DISPROPORTIONATION OF UNSYMMETRICAL ANHYDRIDES The instability of unsymmetrical anhydrides was recognized quite early. Autenrieth (18b) observed that his mixed acetic anhydrides partially decomposed on distillation to acetic anhydride and the other acid anhydride. Because of the in- stability of unsymmetrical anhydrides, their existence was doubted at first by many. (Elemental analysis of an unsymmetrical anhydride yields the same result as an analysis of an equimolar mixture of the two simple anhydrides.) bility is due to disproportionation; cal and the simple anhydrides: The insta- an equilibrium exists between the unsymmetri- -92 R COOCOR2 (R1 CO)2 0 + (R2 CO) 2O (6) Doubt of the existence of unsymmetrical anhydrides disappeared with the isolation of some higher unsymmetrical anhydrides which are solids at ordinary temperatures (40). It was also shown that some liquid unsymmetrical anhydrides could be puri- fied. Autenrieth and Thomae (18 c) distilled acetic isovaleric anhydride without decomposition. The tendency of an unsymmetrical anhydride to disproportionate into the twocomponent simple anhydrides often makes isolation impossible or at least difficult. This behavior greatly complicates the purification and determination of physical constants and frequently requires immediate use after preparation. When the sizes of the two alkyl or aryl groups are decidedly different, unsymmetrical anhydrides tend to disproportionate even on standing at room temperature (28). Zeavin and Fisher (41) prepared some aromatic unsymmetrical anhydrides by reacting a cold ethereal solution of an acid chloride with an ethereal solution of another acid and pyridine. They observed that the yield was decreased considerably by "hydroly- sis" during the course of the preparation in ether, and to inhibit hydrolysis benzene was later substituted for ether. Zeavin and Fisher noticed that some of the unsymmetrical anhydrides were unstable when heated to their melting points or when concentrated benzene solutions were heated. Hydrolysis would occur due to water in the solvent, but part of the decomposition was probably due to disproportionation. Ralston and Reck (42) prepared several isomeric unsymmetrical anhy- drides of saturated aliphatic monocarboxylic acids (acetic-palmitic, butyricmyristic, caproic-lauric, and caprylic-capric). These unsymmetrical anhydrides are soluble in both polar and nonpolar organic solvents. However, in polar solvents, such as acetone and acetonitrile, solution was attended by disproportionation. This tendency was especially marked where the difference in chain length was appreciable, -10- such as with acetic palmitic anhydride. Ralston and Reck concluded therefore that such anhydrides cannot be satisfactorily crystallized from polar organic solvents. They found that nonpolar solvents were suitable for recrystallization if prolonged heating was not necessary. In the case of liquid unsymmetrical anhydrides, flash distillation is the usual method of purification. By this procedure several of the acyl trifluoroacetates, including benzoyl trifluoroacetate, have been isolated (17, 43). The mechanism of the formation of bis (trimellitic anhydride) anhydride from TMA and acetic anhydride (12) is now clear. The initial reaction must have con- sisted of the formation of the unsymmetrical anhydride of TMA and acetic anhydride: 0 0 CC 0o\0 O COOH /C---OOCOCH3 o\ xc + (CH3CO)20 X° +CH3COOH (7) Distilling off the acetic acid and excess acetic anhydride followed by additional heating led to disproportionation and formation of the trianhydride (the simple anhydride of TMA) and acetic anhydride: &Coc^ °\ 20'~ oI CoC00H 3 ,,,,,% I O/ .. ooo / ++ (cOC0. (CH3Co)2 0 (8) -11- HYDROLYSIS OF UNSYMMETRICAL ANHYDRIDES Most unsymmetrical anhydrides are extremely reactive compounds. Another factor besides disproportionation which makes their preparation and purification The presence of moisture in solvents or as water vapor difficult is hydrolysis. can lead to the formation of the component acids. Berliner and Altschul (44) studied the hydrolysis of substituted benzoic anhydrides. The action of the water molecule on the carbonyl carbon, as shown in Reaction (9), followed by the formation of a new covalent bond is affected by the presence of substituents. The reaction, which follows the Hammett equation, is favored by electron-withdrawing groups and is impeded by electron-donating groups, and therefore involves a Unsymmetrical anhydrides nucleophilic attack as the rate-determining step (44). are HO- --- - OCR H-OH R H 6 ° t R HO-C-OCR 6- HOCR 0 rapidly hydrolyzed by water, and the rate increases with greater differences in chain length of the component acids (42). The hydrolysis of acetic formic anhy- dride proceeds one hundred times faster than the hydrolysis of acetic anhydride (45). A study of the hydrolysis of acetic propionic anhydride (40) helped verify the actual existence of unsymmetrical anhydrides. The anhydride was shown to be homogeneous as the hydrolysis study indicated the reaction was of the first order. If the substance investigated were a mixture of the two simple anhydrides, the first order rate coefficients would have decreased with time due to the different rates of hydrolysis of the two simple anhydrides. -12- REACTIONS OF UNSYMMETRICAL ANHYDRIDES When an unsymmetrical anhydride is reacted with an alcohol, esters of either acid may be produced: R1 COOCOR2 + R3OH -- RCOOR R1COOCOR 2 + R3OH R1COOH + R2 COOH (10) OOR R (11) There is the third possibility that esters of both acids may be formed leading to a complex mixture of two acids and two esters. Therefore, the reaction would normally be of little value unless specific for the formation of one ester. One exception of note occurred when various unsymmetrical anhydrides were reacted with cellulose leading to mixed esters of cellulose (46). Several investigations have been carried out on the esterification products of unsymmetrical anhydrides. Some of the studies of products obtained are mentioned below, along with a discussion on the reaction mechanisms of unsymmetrical anhydrides. Previous Investigations on the Esterification of Unsymmetrical Anhydrides The early investigations by Autenrieth were concerned with unsymmetrical anhydrides which gave mixtures of esters. When acetic benzoic anhydride was reacted with an alcohol, both benzoate and acetate were obtained (18a). A similar study by Rollett and Scholz (47) indicated that the yield of ethyl benzoate at 70°C. was 50%; the yield increased at higher temperatures. This study contrasts with recent results whereby acetic benzoic anhydride in boiling ethanol led to the recovery of 90% ethyl acetate (48). Autenrieth also studied the esterification of acetic isovaleric anhydride (18b). The major product after refluxing with phenol was phenyl acetate. ing with ethanol led to a mixture of ethyl esters. Staudinger (30) reacted Reflux- -13- diphenylacetic benzoic anhydride with an alcohol and obtained mainly the diphenylacetate ester. The reaction of acetic propionic anhydride led to a 2:1 ratio of acetate ester to propionate ester (49). Where the two acids differ greatly in acid strength, the ester of the stronger acid is often obtained. This was indicated by the reaction of acetic chloroacetic anhydride with ethanol, where ethyl chloroacetate in yields up to .98.5 was obtained (50-51). This would also agree with the results of Kahn (52) who obtained the p- nitrobenzoate when alcohol (presumably ethanol) was reacted with benzoic p-nitrobenzoic anhydride. With acids of similar strength, such as benzoic p-isopropyl- benzoic anhydride, Kahn obtained a mixture of esters. Although it has been stated that the strengths of the component acids determine the products formed, this rule does not always hold true. It has been noted that some unsymmetrical anhydrides react with an alcohol or amine to yield the ester or amide derived from the weaker acid as the major product. When Kahn (52) reacted benzoic mesitoic anhydride with ethanol he obtained ethyl mesitoate almost exclusively, i.e., the ester of the weaker and sterically hindered acid. This was also noted in the ammonolysis of benzoic acetic anhydride (26, 27a) and of acetic trichloroacetic anhydride (53) where the acetyl derivative greatly predominated. Emery and Gold (54) also demon- strated that the competition of the acylation products is greatly influenced by the nature of the solvent employed. The reaction of acetic chloroacetic anhydride and aniline carried out in benzene led to the formation of 86% chloroacetanilide. With more polar solvents, however, acetylation was the primary reaction. In 50% acetone-water, 72% acetanilide was obtained. The Mechanism of Acylation by Unsymmetrical Anhydrides In some reaction systems, the products obtained may be greatly dependent on the position of the equilibrium of the disproportionation reaction illustrated in -14- Reaction (6). Assuming that the unsymmetrical anhydride predominates, the un- catalyzed reaction could be explained by two different reaction mechanisms (55-56). The first category of reactions would be that type involving a bimolecular displacement of the SN2 type: 0 0 R 0 I1 11 -, 5 . IN -11 BH+R C-O-CR2- BH---C ---OCR2- + R1 COHB +R2COO - R1COB+ R2COOH (12) 0 The second type (56) is that where the anhydride undergoes preliminary ionization and which can be compared to an SN1 reaction: R 1 COOCOR2 slow · ' R1 CO+ R CO+ + BH fast R1COHB + 1 %'1 + R2COO _ fast fast' + R2 COO- (13) R1COHB+ (14) BC R COB + R2 COOH (15) The bimolecular reaction is probably the one usually in effect. This mechanism predicts the results of reaction of most unsymmetrical anhydrides. This mechanism would favor the formation of derivatives of the stronger acid, as nucleophilic attack would occur preferentially at the carbonyl group which was most deficient in electrons. Thus, in Reaction (12), reaction with the molecular anhydride, R1 COB would be the major product if R1 were more strongly electron attracting than R2 . In considering the SN1 mechanism, Reactions (13) to (15), it would be expected that heterolytic fission would be more likely with an unsymmetrical molecule, and that ionization would give the normal negative ion of the stronger acid and the acylium ion of the weaker acid, rather than the reverse, so that the over-all reaction would result in the acyl derivative of the weaker acid being prepared (56). -15- The above two mechanisms can be used to interpret most of the acylation reactions with unsymmetrical anhydrides. Under conditions favoring reaction with the molecular anhydride, the ester of the stronger acid will be prepared. Under conditions favoring ionization, the ester of the weaker acid will be prepared. The predominant preparation of the acyl derivatives derived from the stronger acid is consistent with the SN2 mechanism (56). Instances where derivatives of weaker acids are obtained may not always be explained by the SN1 mechanism. One such instance is the reaction of acetic chloroacetic anhydride with 2,4-dichloroacetanilide almost exclusively. mechanism was still in effect. Emery and Gold (53) concluded that a bimolecular They based their reasoning on the characteristic features of an ionization mechanism for acylation. If the SN1 mechanism were in effect, then there would be a rate-determining step (ionization) which would be independent of the nature of the second reactant. However, the velocity of acylation depends strongly on the nature of the amine employed and, therefore, the existence of a common rate-determining step could be ruled out. Furthermore, the chloroacetylation ratio (chloroacetylation:acetylation) does not depend strongly on the nature and concentration of the amine, as it should if one of the acetylation reactions occurred by an ionic mechanism. In addition, their results did not indicate the existence of specific and common ion intervention by acetate and chloroacetate anions, as would be expected on the basis of an ionization reaction mechanism (57). Another argument against the ionization mechanism is that ioniza- tion would be very unlikely in a nonpolar solvent such as benzene. Emery and Gold (53) studied acylation reactions in benzene of the series of unsymmetrical anhydrides:acetic chloroacetic, acetic dichloroacetic, and acetic trichloroacetic. As mentioned above, they concluded that a bimolecular mechanism was operative and that the activating inductive effect of chlorine substitution was -16- opposed by a steric factor. a CH3 COOAc 1 For the series: a C1CH COOAc 6.1 a C12CHCOOAc 2.3 a Cl CCOOAc 0.08 the actual chloroacetylation ratios in benzene are presented. The steric effect of successive chlorine substitution on the bimolecular reaction at (a) would exert a retarding influence, but electron withdrawal by the chlorine atoms would exert an opposite effect. The electronic effect is then the controlling factor at the beginning of the series and the steric effect is controlling for the highly substituted members. According to Tedder (56), similar arguments can be applied to some of the other anomalous reactions. However, the reaction of benzoic mesitoic anhydride to yield the ester of the weaker acid (52) cannot be explained in terms of steric hindrance, as the ester of the more sterically hindered acid is obtained. In this case, the reaction must proceed by the ionization mechanism, particularly since the 2,4,6-trimethylbenzoylium cation is known to be quite stable (58). This would be analogous to the esterification of mesitoic acid catalyzed by concentrated sulfuric acid, which is also said to proceed through the 2,4,6-trimethylbenzoylium ion (59). Trifluoroacetic Anhydride as an Esterification Promoter Trifluorcacetic anhydride, hereafter referred to as TFAA, will be considered separately from other anhydrides due to its wide use as an esterification promoter. TFAA has been employed in several studies to promote the acetylation of cellulose (60-63). TFAA has also been used to promote the esterification of cellulose with some aromatic acids, such as the benzoate (60, 61) and the phenoxyacetate, P-benzoyl acrylate, phenylmercaptoacetate, p-tert-butylbenzoate, cinnamate, p-methoxyphenyl- -17acetate, and the p-isopropyl phenylacetate (64). It has been shown to be a very powerful esterification promoter as it even catalyzed the formation of acrylates and methacrylates of the 1,1-dihydroperfluoroalkyl alcohols, which are very difficult to esterify (65). Bourne, et al. have suggested (61) that in the reactions promoted by TFAA, an unsymmetrical anhydride is formed. The acyl trifluoroacetate would be formed by (20): RCOOH + (CF3C0)20 RCOOCOCF - + CF3COOH (16) Bourne hypothesized that the unsymmetrical anhydride then ionizes to a slight extent into acylium (RCO + ) and trifluoroacetate (CF3COO-) ions, the acylium ion being the principal acylating species: RCOOCOCF3 (RCO)+ + (CF 3 COO) - _· RCO + + CF3COO- + R'OH -- (17) RCOOR' + CF3 COOH (18) The alternative ionization into (CF3CO+) and (RCOO-) ions would be improbable due to the strong electronegative character of fluorine. Morgan (66) determined the freezing point depression when a small amount of TFAA was added to glacial acetic acid. His data indicated that each TFAA molecule gave rise to two other molecules, which is in agreement with Equation (16) if it may be assumed that the degree of ionization of the unsymmetrical anhydride is small. This supposedly eliminated the possibilities of no reaction and the forma- tion of acetic anhydride, which would require that each TFAA molecule gave rise to three other molecules by the reaction: (cF3Co)0 + 2 CFHC C O 2 (CH0) (CC) 2 CCOOH (19) -18- However, Bourne, and others (67), in repeating this work disagreed with Morgan. Bourne's work indicated that in a large excess of acetic acid, acetic anhydride would be favored rather than acetyl trifluoroacetate. However, in equimolar quantities of acetic acid and TFAA, the formation of acetyl trifluoroacetate would be favored. Emmons, et al. (43) followed the reaction of benzoic acid and TFAA in n-butyl ether and in acetonitrile with infrared spectra, and indicated that the formation of the unsymmetrical anhydride was almost quantitative. Bourne, et al. showed that a composite curve obtained by addition of the optical densities of the two solutions of acetic anhydride and TFAA in carbon tetrachloride, was almost identical with the spectrum of a mixture of the two anhydrides immediately after mixing (68). However, a new spectrum obtained after an hour showed the absorp- tion bands due to the formation of acetyl trifluoroacetate and a decrease in absorption due to the symmetrical anhydrides. As mentioned above, some of the acyl trifluoroacetates have been isolated and purified (17, 20, 43). Randles, et al. (69) attempted to explain the reactivity of the medium of TFAA and acetic acid as an acetylating agent by electrical conductivity determinations. TFAA has an extremely low specific conductivity, much lower than that of trifluoroacetic acid. However, the conductivity of a dilute solution of TFAA in acetic acid was greater than that of a solution having twice this concentration of trifluoroacetic acid in acetic acid. This has been taken as support for the hypothesis of the formation and ionization of an unsymmetrical anhydride. It would be expected that an unsymmetrical anhydride could ionize more readily than one of a symmetrical anhydride, particularly when the two component acids differ widely in strength. Randles agreed that Bourne's postulated ionization mechanism was the most probable explanation. -19Some indirect evidence for the preliminary ionization of the acetyl trifluoroacetate before acetylation was obtained by the addition of sodium acetate to acetic acid, TFAA, and an alcohol (70). This addition greatly reduced the acetylating power and both acetate and trifluoroacetate esters were then obtained. Bourne suggested that the base increased the concentration of the trifluoroacetate anion, thus decreasing the concentration of the reactive cation (CH3CO+) in favor of the molecular anhydrides CH3COOCOCF 3 and TFAA, both of which are capable of promoting trifluoroacetylation. If esterifications promoted by TFAA do proceed through the formation of an unsymmetrical anhydride followed by the ionization reaction mechanism, then TFAA should promote esterification of sterically hindered acids in the same manner as concentrated sulfuric acid catalyzes the esterification of mesitoic acid (59). Reed (71) demonstrated that TFAA was effective in the esterification of certain sterically hindered acids, such as trimethylacetic, 2,6-dimethyl benzoic, and 2,6-dimethyl cyclohexane carboxylic acid. These acids are unreactive by the usual acid-catalyzed esterification procedures. Although acylation takes place under most conditions, there are a few cases in which trifluoroacetylation may also occur. Bourne (20) attempted to explain this by suggesting that in the molecular form the unsymmetrical anhydride may function both as an acylating and trifluoroacetylating agent. Bourne (72) found that the unsymmetrical anhydride benzoyl trifluoroacetate showed a greater tendency to form trifluoroacetate esters than acetyl trifluoroacetate. Bourne suggested that this was due to resonance stabilization of the benzoylium cation, creating a lowering of the density of the positive charge on the carbonyl group. This would result in a slowing down of the reaction of the cation with hydroxyl groups, thus allowing reaction of the molecular anhydride to assume a greater relative importance. -20- In addition, the greater size of the phenyl group would also mean that steric factors would not favor benzoylation as much as trifluoroacetylation. However, the proportion of benzoate ester could be greatly increased by carrying out the reaction in trifluoroacetic acid or by reacting the unsymmetrical anhydride with phenols or secondary alcohols instead of primary alcohols. It is sometimes difficult to determine the extent of trifluoroacetylation since the esters readily hydrolyze in the presence of water, particularly the fluorinated esters of polyhydroxy compounds (73). In esters of this type the carbonyl carbon is very susceptible to nucleophilic attack due to the strong inductive effect exerted by the perfluorinated group a to the carbonyl (74). The electron-withdrawing CF3 - group decreases the ability of an ester to accept protons and increases the tendency of a water molecule to combine with the carbonyl group (75). However, if the perfluoroester is insoluble in water, it is not as easily hydrolyzed (76). Bourne (72) summarized by stating that acyl trifluoroacetates react with alcohols to give trifluoroacetyl and acyl esters in proportions depending on the nature of the unsymmetrical anhydrides and the hydroxyl compounds; and that acyl esters usually predominate. The yield of acyl ester is usually increased by the presence of trifluoroacetic acid and solvents of high ionizing ability and decreased by the addition of sodium trifluoroacetate. The influence of trifluoro- acetic acid would be due partly to its ability to solvate the unsymmetrical anhydride and partly to acid catalysis. Functioning as an acid catalyst, trifluoro- acetic acid would promote the ionization resulting in acylation by the weaker acid group (56): CF3COOCOR + H + - CF3COOCR OH+ - CF COOH + RCO+ (20) -21- Using a solvent of high ionizing powers, the separation of ions should be assisted (77). The addition of sodium trifluoroacetate, by the common-ion effect, would suppress the ionization of the unsymmetrical anhydride. -22- PRESENTATION AND APPROACH TO THE PROBLEM When TMA is heated with an alcohol, the cyclic anhydride ring is opened resulting in a mixture of monoesters. Further reaction to triesters and poly- esters can be obtained through the use of a catalyst such as sulfuric acid. Since TMA possesses a free carboxyl group, it seemed possible to pick reaction conditions such that an unsymmetrical anhydride of TMA and another acid could be formed: (RCO)2 0 + 0 HOOC -C 0 RCOOH + o 0 0 (21) 0 RCOC /-C/o These reactive unsymmetrical anhydrides, which would be much more reactive than cyclic anhydrides, could well be very important intermediates for specific esterification. An unsymmetrical anhydride of TMA and another acid could react with an alcohol to give esters of either or both carboxylic compounds depending on the nature of the acids and alcohol involved and on steric factors. 4 RCOOC /0 + R2 OH--> RCOOR2 + -CO - H HOOC >RCOOH + R2 0° C 0 0 C ho (22) -23- It was of interest in this thesis study to prepare the unsymmetrical anhydrides and investigate the above esterifications. The second reaction, the preparation of the 4-carboalkoxy phthalic anhydrides, would be of particular interest. The initial study would be performed on the TMA-TFAA reaction system, partly because of the work of Bourne which suggested that the unsymmetrical anhydride should be formed readily. Most reaction systems of TFAA and another carboxylic acid had led to esters of the latter acid. be quite stable with this type of reaction. The cyclic anhydride ring of TMA should Tedder (56) has indicated that dibasic acids are not readily esterified in the presence of TFAA if they form stable cyclic anhydrides. As an example, no esterification occurred when phthalic acid was treated with p-nitrobenzyl alcohol and TFAA, yet it was possible to obtain a 69% yield of p-nitrobenzyl benzoate under corresponding conditions. It will also be remembered, that one method of preparing cyclic anhydrides is by refluxing the diacid in a simpler anhydride, such as acetic anhydride (78-80). This further suggests the stability of cyclic anhydride rings under the above conditions. The approach to the problem included the preparation of the unsymmetrical anhydrides (the trifluoroacetyl, acetyl, and acid chloride derivatives) and the simple anhydride of TMA, bis(trimellitic anhydride) anhydride. The anhydride derivatives would be characterized, and their stability and esterification properties would be investigated. Simple alcohols were chosen for the esterification reactions, with the objective of obtaining monoesters. obtain polyesters from the substituted derivatives. No attempt was made to -24- PROCEDURES AND RESULTS On the following pages, descriptions are given of the preparations, purifications, analyses, and esterifications of four anhydride derivatives of TMA: the trifluoroacetyl and acetyl unsymmetrical anhydrides of TMA, the acid chloride of TMA, and the simple anhydride of TMA. Specific analytical procedures such as anhydride equivalent, fluorine content, and molecular weight are presented in the Appendix. The infrared spectra were obtained on KBr disks unless otherwise indi- cated. THE TRIMELLITIC ANHYDRIDE-TRIFLUOROACETIC ANHYDRIDE REACTION SYSTEM The unsymmetrical anhydride of TMA and TFAA was isolated, purified, and characterized. The stability of the anhydride was considered and a number of esterification reactions with various alcohols were conducted. PREPARATION OF THE UNSYMMETRICAL ANHYDRIDE OF TMA AND TFAA It was determined that the crude unsymmetrical anhydride could be obtained easily in high yield by refluxing the two reactants. (0.026 mole;) and 30 g. TFAA (0.143 mole') For example, 5 g. TMA were refluxed five hours. The hot solution was filtered and the filtrate solidified on cooling to room temperature. After drying under vacuum, 5.61 g. were obtained (74.8% yield). On heating above the melting point (91-94 ° ), the evolution of a gas was followed by solidification and remelting at a higher temperature (about 220° ). The unsymmetrical anhydride could also be obtained by reaction at room temperature or under pressure in a glass tube, but this was more time-consuming and did not always result in a product as pure as that obtained from the reaction conducted at reflux temperature. -25- PURIFICATION AND ANALYSIS OF THE UNSYMMETRICAL ANHYDRIDE OF TMA AND TFAA Great difficulty was experienced in attempts to purify the unsymmetrical anhydride of TMA and TFAA, due to its tendency for disproportionation and its great affinity for water, resulting in hydrolysis of the linear anhydride group. As the anhydride decomposed during an attempt at sublimation, it seemed that recrystallization would be the most promising technique. Other purification tech- niques would be severely hindered due to the solubility properties and due to the necessary exclusion of such reactive chemicals as water, alcohols, and amines. The unsymmetrical anhydride was finally recrystallized successfully from benzene, but not before numerous other solvents had been investigated and found unsatisfactory (nitroethane, methyl ethyl ketone, carbon tetrachloride, petroleum ether, o-dichlorobenzene, anisole, acetone, toluene, ether, and cyclohexane). The crude unsymmetrical anhydride was prepared by refluxing a solution of 5.0 g. TMA (0.026 mole) in 30 g. TFAA (0.143 mole). On cooling, the solidified product was filtered off and dried, m.p. 93-98, 218-220° . The product was re- crystallized by solution in benzene at room temperature, followed by filtration, concentration of the filtrate, and standing at room temperature. lized product had m.p. 92-95, 222 ° , yield: 1.8 g. The recrystal- When analyzed for anhydride equivalent with methanolic morpholine (see Appendix I for this procedure) a value much higher than theoretical was obtained. The determination of neutralization equivalent with methanolic sodium hydroxide also led to a high value. Since dilute standard solutions were employed for these determinations, it was believed possible that the methanol was reacting with the unsymmetrical anhydride. Experimentation indicated that morpholine in acetone could be used for the determination of anhydride equivalent and aqueous sodium hydroxide could be used for the determination -26- of neutralization equivalents. 152 (theoretical: 144.067). obtained, m.p. 92-95, 222°. The anhydride equivalent determined in acetone was After the second recrystallization, 1.11 g. were The neutralization equivalent was 74.4 (theoretical: 72.03) and the anhydride equivalent determined in acetone was 156 (theoretical: 144.067). After the third recrystallization, m.p. 94-96, 223 ° , the sample had a neutralization equivalent of 73.9 (theoretical: determined in acetone was 150 (theoretical: (theoretical: 288.13). 72.03). The anhydride equivalent 144.067) while in methanol it was 277 To eliminate the possibility of a polyanhydride, the molecular weight was determined by the freezing point depression of benzene. Appendix II.) The determined molecular weight was 274 + 3 (theoretical: (See 288.13). A sample of the purified unsymmetrical anhydride was submitted to the Geller Laboratories for carbon, hydrogen, and fluorine analysis. The analysis obtained (56.53% C, 1.93% H, and 1.68% F) was quite different from the theoretical values (45.85% C, 1.05% H, and 19.78% F). The unsymmetrical anhydride appeared to be too unstable to be analyzed by an outside laboratory due to the long delay necessary before analysis. Since some form of elemental analysis was still desirable for characterization of the compound, it was decided to determine the fluorine content immediately after the preparation and purification of the unsymmetrical anhydride. The method employed was an oxygen combustion followed by titration with CeC13 (see Appendix III). A satisfactory analysis was obtained: 19.3, 19.5% F (theoretical: 19.78% F). Studies of the infrared spectra indicated carbonyl absorption bands probably due to the cyclic anhydride ring at 1870 and 1778 cm.-1 1860 and 1778 cm. ). As expected with cyclic anhydrides, the lower frequency band had the greater absorption. linear anhydride. (TMA exhibits bands at Bands at 1838 and 1765 cm. were probably due to the The former band had slightly greater intensity. The strongest -27- carbonyl band of the unsymmetrical anhydride of TFAA and benzoic anhydride is at 1835 cm.- (43). The infrared spectrum indicated that the anhydride was not completely pure, however, as there was weak absorption at 1710 cm.-1 (carboxyl). DISPROPORTIONATION OF THE UNSYMMETRICAL ANHYDRIDE OF TMA AND TFAA The unusual melting point of the unsymmetrical anhydride of TMA and TFAA suggested that the product decomposed under the influence of heat. This decom- position should consist of disproportionation to the simple anhydrides, TFAA and bis(trimellitic anhydride) anhydride,which is actually a trianhydride. A sample of the mixed anhydride of TMA and TFAA was prepared to test the hypothesis that its decomposition product was the trianhydride. anhydride (2.91 g.) The unsymmetrical was heated in an evaporating dish on a hot plate. melted and then bubbled. The odor of TFAA was noticeable. ified and was placed in a vacuum desiccator to cool. The sample The sample then solid- The product obtained (2.1 g., 113% yield based on the trianhydride) had m.p. 210-224°. The seemingly inconsis- tent yield figure could be due to a small amount of TMA in the starting material. The product was recrystallized four times from 1,2-dichloroethane, m.p. 223-228°, and mixed m.p. with the trianhydride, 224-228° . (theoretical: 122.08). The anhydride equivalent was 126 An infrared spectrum indicated carbonyl absorption at 1861, 1800, 1777, and 1738 cm. . TMA absorbs at 1860 and 1778 cm. , correspond- ing to the first and third bands and accounting for the cyclic anhydride. The linear anhydride could be similar to benzoyl benzoate, which absorbs at 1789 and 1727 cm.- 1 (81), which are at slightly lower frequencies than the second and fourth bands. There was no indication of carboxyl absorption. It was also demonstrated that the trianhydride could be obtained directly by continued treatment of TMA in TFAA. TMA (4.0 g., 0.0208 mole) was treated with -28- TFAA (37.5 g., 0.178 mole) in an all glass tube. The mixture was heated to solution and cooled to room temperature a number of times over a period of several days. On final cooling, 2.55 g. of the trianhydride separated, m.p. 218-222° (yield: 62%). On recrystallization from 1,2-dichloroethane, 1.8 g. were recovered, m.p. 222-226° and anhydride equivalent: 129 (theoretical: 122.08). The second crop to separate from the TFAA solution was the unsymmetrical anhydride, m.p. 96.5-98.5, 219 ° and yield: .0.69 g. (11.5%). While attempting to recrystallize the unsymmetrical anhydride, a peculiar solvent effect was noted. In certain solvents the unsymmetrical anhydride dis- proportionated to the trianhydride, even at room temperature or below. A sample of the unsymmetrical anhydride was dissolved in nitroethane at room temperature. After two recrystallizations the trianhydride was recovered, m.p. 216-221° . Cold anhydrous ether was added to a second sample of the unsymmetrical anhydride. product dissolved and then, within minutes, crystallization took place. crystallized product was also the trianhydride, m.p. 218-221 ° The This . HYDROLYSIS OF THE UNSYMMETRICAL ANHYDRIDE OF TMA AND TFAA The unsymmetrical anhydride of TMA and TFAA has a great affinity for water. (Partial hydrolysis would result in TMA and trifluoroacetic acid.) was observed in early attempts to recrystallize the anhydride. This behavior In some cases, small amounts of moisture present in the solvent or absorbed by the solution must have been responsible for hydrolysis. dissolved in carbon tetrachloride. A sample of the unsymmetrical anhydride was The product which recrystallized from the solu- tion had a m.p. of 166-169 ° and an anhydride equivalent of 204 (values for TMA: 168 ° and 192). Attempts to recrystallize from toluene, cyclohexane, petroleum ether, and o-dichlorobenzene also led to products with melting points higher than the unsymmetrical anhydride, indicating that hydrolysis had taken place. -29- The affinity of the unsymmetrical anhydride for water or water vapor was best exhibited by a series of infrared spectra. A sample of the crude unsymmetrical anhydride (m.p. 91-93, 223°) was pressed into a potassium bromide pellet and a spectrum was obtained. However, the spectrum had several similarities to TMA. as compared The main carbonyl absorption bands were at 1870, 1785, and 1712 cm. with 1860, 1778, and 1717 cm. for TMA. The acid band was not as strong as with TMA, but it was not possible to pick out the two pairs of anhydride bands expected with a dianhydride of this type. Moisture adsorbed by the potassium bromide or adsorbed during the grinding process and the formation of the pellet, must have reacted with some of the unsymmetrical anhydride resulting in the formation of some TMA. A second spectrum was obtained on a nujol mull of the sample. Carboxyl absorption be expected, there was very little hydrolysis in this case. was exhibited at 1710 cm. but with much less intensity. As might The two pairs of anhy- dride carbonyl bands were present at 1874, 1840, 1788, and 1767 cm. -1 . The un- symmetrical anhydride was recrystallized four times from benzene and infrared spectra were obtained after the first, second, and fourth recrystallizations. Anhydride carbonyl bands of the recrystallized product were at 1870, 1838, 1778, and 1765 cm. -1 Although the carboxyl band at 1710 cm. -1 the band did not disappear on repeated recrystallization. decreased in intensity, A small degree of hydrolysis always appeared to take place in the time interval between recrystallization and completion of analysis. ESTERIFICATION OF THE UNSYMMETRICAL ANHYDRIDE OF TMA AND TFAA A number of esterifications were conducted between equimolar amounts of the unsymmetrical anhydride and various alcohols under different reaction conditions. The reactions are summarized in Table I, while the experimental details are presented in Appendix IV. TABLE I ESTERIFICATION REACTIONS OF THE UNSYMMETRICAL ANHYDRIDE OF TMA AND TFAA Solvent Alcohol Temp., °C. Products None Phenol 75 None Butanol-1 75 TMA n-butyl trifluoroacetate None Methanol 25 TMA methyl trifluoroacetate Acetone Butanol-l 25 n-butyl trifluoroacetate Acetone Phenol 25 TMA trianhydride Acetone o-Cresol 25 TMA trianhydride Acetone None 25 trianhydride Trifluoroacetic acid Methanol 25 TMA Trifluoroacetic acid Butanol-2 25 TMA Trifluoroacetic acid Butanol-2 85 TMA Trifluoroacetic acid Capryl 25 TMA Trifluoroacetic acid Phenol 83 TMA monophenyl trimellitate 25 TMA n-butyl trifluoroacetate Ether Butanol-1 TMA CCl4 Phenol 75 TMA o-Dichlorobenzene Butanol-2 25 TMA 1,2-Dichloroethane Butanol-2 25 TMA -31In all cases, except the reactions in acetone, TMA was recovered in very high yields (usually 80 to 90%). The trifluoroacetate esters were also identified in some of the esterifications. None of the ester anhydrides of TMA were isolated, and they could have been formed in only very small quantities. The esterifications in acetone led to the isolation of both TMA and the trianhydride, bis(trimellitic' anhydride) anhydride. Three additional esterifications were conducted with alcohols in greater than equimolar quantities (methanol, butanol-l, and butanol-2). With excess alcohol, the cyclic anhydride ring of TMA is opened, leading to mixtures of monoesters. THE UNSYMMETRICAL ANHYDRIDE OF TRIMELLITIC AND ACETIC ANHYDRIDES The unsymmetrical anhydride of TMA and acetic anhydride was prepared, purified, and analyzed. A few brief reactions with methanol were conducted to determine the possibility of obtaining 4-carbomethoxy phthalic anhydride. This ester anhydride was not obtained. PREPARATION OF THE UNSYMMETRICAL ANHYDRIDE The unsymmetrical anhydride of TMA and acetic anhydride was first prepared in the following manner. TMA (10 g., an excess of acetic anhydride. 0.0522 mole) was refluxed for 2.5 hours in On cooling, there was no crystallization, so the mixture was distilled to remove acetic acid and some of the acetic anhydride. mixture was then cooled in an ice-salt water bath for two hours. The The fine white precipitate was filtered, washed withacetic anhydride, and washed with benzene. The dried product melted at 77-79° (TMA melts at 168 ° ) and must have been the unsymmetrical anhydride. cal: This material had an anhydride equivalent of 118 (theoreti- 117.1). An infrared spectrum indicated that this dianhydride had the anhydride -32- carbonyl bands of TMA at 1860 and 1778 cm. 1 and another carbonyl pair at 1818 and 1733 cm. -1 , probably due to the unsymmetrical anhydride (the carbonyl bands of acetic anhydride are at 1825 and 1748 cm.-). Later attempts to prepare the unsymmetrical anhydride by this procedure were not quite as successful. The crude product was never obtained in as pure a form as in the original preparation. In the first attempt to repeat this reaction, 15 g. (0.0782 mole) TMA were refluxed 2.5 hours with 100 ml. acetic anhydride (1.06 moles). The acetic acid and some excess acetic anhydride (60 ml.) were then distilled from the solution. No precipitation occurred on cooling. Alternate steps of concentration and cooling resulted ultimately in a dark sirup with no precipitation. Attempts to cause precipitation by the addition of benzene, carbon tetrachloride, o-dichlorobenzene, and p-dioxane were unsuccessful. After refrigera- tion for several days, however, the sirup solidified to give 18.6 g. of an impure product which melted over a wide range (77-160°). In the next attempt to prepare the unsymmetrical anhydride, 10 g. TMA (0.0522 mole) and 75 ml. acetic anhydride (0.794 mole) were refluxed three hours, followed by the distillation of 36 ml. (38.4 g.) from the solution. refrigeration. In this case, a white powder (1.1 g.) separated on However, this was also impure, m.p. 78-125 °. Diluting the reactant mixture with acetic acid in another preparation led to recovery of unreacted TMA. A purer form of the unsymmetrical anhydride can be obtained, however, by cooling the reactant mixture in acetone-dry ice and allowing it to warm slowly to about -50° . The crude product obtained by this procedure was never in quite as pure a form as in the original preparation. PURIFICATION AND ANALYSIS OF THE UNSYMMETRICAL ANHYDRIDE The unsymmetrical anhydride was prepared for purification and analysis. (10 g., 0.0522 mole) and acetic anhydride (81.08 g., 0.794 mole) were refluxed TMA -33three hours. After the removal of 38.4 g. by distillation, refrigeration induced the precipitation of 2.6 g. material which was probably a mixture of TMA and the unsymmetrical anhydride. On cooling the reactant mixture to -79° in acetone-dry ice and allowing it to warm slowly, the crude unsymmetrical anhydride separated, m.p. 71-80 ° and yield 2.11 g. This product was employed to investigate various possible purification techniques. The results indicated that the unsymmetrical anhydride of TMA and acetic anhydride could be purified by recrystallization from benzene or by extraction of the TMA impurity with cold ether. Continued cooling of the reactant mixture in acetone-dry ice led to the further isolation of 0.87 g., m.p. 69-115 ° . m.p. 79-83 °. Benzene extraction and recrystallization led to a product of Additional recrystallization from benzene yielded the purest form of the unsymmetrical anhydride of TMA and acetic anhydride yet obtained. product had a m.p. of 84-86°. This The anhydride equivalent was 121 (theoretical: 117.1), while the neutralization equivalent was 59.3 (theoretical: 58.54), and the molecular weight was 225 + 10 (theoretical: 234.16). The infrared spectrum was consistent with the proposed structure (see Fig. 1). There was no carboxyl absorption. cm. -1 Carbonyl absorption due to the cyclic anhydride was at 1863 and 1778 (TMA has bands at 1860 and 1778 cm. 1 .) to the linear anhydride was at 1812 and 1732 cm. could be due to -CH3. Carbonyl absorption attributed . A medium band at 1372 cm. This band is not present in TMA or its anhydride. Duplicate carbon and hydrogen analyses were obtained on the sample by the Geller Laboratories. Carbon and hydrogen content along with the oxygen content (by difference) are presented in Table II with the theoretical values for the unsymmetrical anhydride of TMA and acetic anhydride. Attempts to recrystallize the unsymmetrical anhydride from the crude material with the solvents acetonitrile, p-dioxane, acetone, acetic anhydride, o-dichlorobenzene, vinyl trichloride, and 1,2-dichloroethane were unsuccessful. An attempt -134- ) 0 P 0 0 0 Go 0 0 0 0 I cIx co U- 0 CL) U C (D z a ci: Lic I D cw -35to purify by dissolving the sample in acetone at room temperature followed by drying over phosphorus pentoxide also yielded a mixture, Sublimation attempts at 70° and 1-2 mm. did not lead to the pure unsymmetrical anhydride. Recrystallization from benzene or ether extraction were the only successful methods. TABLE II ELEMENTAL ANALYSIS OF 4-ACETYL TMA C, % H, % (1) 56.06 2.72 (2) 55.71 2.73 Av. 55.89 2.73 41,38 Theoretical 56.42 2.58 41.00 Sample 0, % REACTION OF THE UNSYMMETRICAL ANHYDRIDE WITH METHANOL Three reactions were carried out between methanol and the unsymmetrical anhydride of TMA and acetic anhydride to determine the possibility of obtaining 4carbomethoxy-phthalic anhydride. However, this methyl ester anhydride was not obtained in pure form. In the first reaction, 0.0490 g. methanol (0.0015 mole) was added to 0.3577 g. of the solid unsymmetrical anhydride (0.0015 mole). sublimation. The product was subjected to After two days at 80 ° and 1-2 mm., only 0.01 g. of sublimate was collected, m.p. 140-155 ° . present in this product. Very little 4-carbomethoxy phthalic anhydride could be The ester, m.p. 94-99 ° (1) or 104-105 ° (2), sublimes much more readily than TMA, m.p. 168 °. Two additional reactions were carried out in benzene. In the first reaction, 1.03 g. of the unsymmetrical anhydride (0.0044 mole) were dissolved in 50 ml. benzene and 0.1426 g. methanol (0.0044 mole) was added. The solution was refluxed for eighteen hours while protected from the atmosphere by a drying tube. After the distillation of 25 ml. solvent, the solution was concentrated to dryness in vacuo at room temperature. completely melt until 145° . The product softened slightly at about 90° but did not This solid was subjected to sublimation. After five days at 80° and 1-2 mm., 0.1 g. sublimate were collected, m.p. 100-145 ° . This product could have contained a small amount of the ester, but the mixture was not separated. This reaction was repeated by refluxing 0.77 g. (0.0033 mole) of the unsymmetrical anhydride and 0.1109 g. (0.0034 mole) methanol in 50 ml. benzene for 24 hours. The sublimate collected after two days (0.03 g.) had m.p. 128-155 ° , and was probably mostly TMA. THE ACID CHLORIDE OF TRIMELLITIC ANHYDRIDE Since the trifluoroacetic and acetic unsymmetrical anhydrides of TMA appeared to react with alcohols to yield primarily the trifluoroacetate or acetate esters plus TMA, it was decided to attempt to obtain an unsymmetrical anhydride which would be more specific for the formation of trimellitate ester anhydrides. The monoacyl chloride of TMA, or phthalic anhydride-4-carboxylic acid chloride, seemed quite promising. To prepare this acyl halide-acid anhydride, a rather mild chlor- inating agent seemed necessary. The reagent would have to react with the carboxyl group without affecting the anhydride group. Previous work discussed earlier suggested that phosphorus pentachloride would be too reactive, but thionyl chloride might be suitable. REACTION OF TMA WITH THIONYL CHLORIDE Refluxing TMA in thionyl chloride until solution was obtained proved successful, after several mild preliminary reactions indicated more drastic conditions were -37necessary. (Reactions at room temperature under vacuum or gentle warming of a mixture of the two reactants led to the recovery of mixtures of products, which appeared to be composed mainly of TMA.) Formation of the Trianhydride Since the anhydride ring did not appear to be affected by thionyl chloride, the reaction was conducted at reflux temperature. TMA (5 g., 0.0262 mole) was heated in 32.7 g. (0.275 mole) thionyl chloride. Hydrogen chloride was evolved during the reaction. Complete solution was obtained after four hours. After an additional two-hour reflux, the solution was cooled, leading to the separation of 0.4 g. white crystals, m.p. 219-223 °. This product was shown to be bis(tri- mellitic anhydride) anhydride, mixed m.p. 220-224 ° and anhydride equivalent: 121 (theoretical: 122.08). Formation of the Acid Chloride of Trimellitic Anhydride The remainder of the solution was vacuum concentrated to dryness leaving 4.4 g., m.p. 63.5-66°. (Yield: 80.o3 based on the formation of the acid chloride). This product gave a strong chlorine test with ethanolic silver nitrate (insoluble in nitric acid), and was soluble in benzene and acetone with limited solubility in water and ethanol. The equivalent weight determined by dissolving the sample in methanol and titrating with sodium hydroxide was 107. The theoretical value on the assumption of the titration of two functional groups would be 105.3. dride equivalent was 123 (theoretical: 210.57). The anhy- Actually this method should not be applied here as the acid chloride interferes by reacting with both methanol and morpholine and provides further interference by releasing hydrogen chloride. An infrared spectrum in potassium bromide indicated carbonyl absorption at 1857, 1779, and 1748 cm.1. -38PURIFICATION AND ANALYSIS OF THE ACID CHLORIDE A sample of the crude acid chloride was dissolved in benzene at room temperature and filtered. On refrigeration, a very small amount of the trianhydride separated, m.p. 221-224 ° . Concentration and continued refrigeration led to the recrystallization of the acid chloride of TMA, m.p. 65-66.5° . The acid chloride had a neutralization equivalent in aqueous solution of 53.5 and a molecular weight of 211 + 4 (theoretical: 52.64 and 210.57). An infrared spectrum in potassium bromide (Fig. 1) indicated carbonyl absorption at 1848 and 1778 cm.-1 (cyclic anhydride) and at 1743 cm. - (acid chloride). A spectrum in nujol indicated no carboxyl or hydroxyl activity, with carbonyl absorption at 1847, 1770, and 1740 -1 cm.. Carbon, hydrogen, and chlorine analyses were obtained by the Geller Laboratories. Their results along with the oxygen content (by difference) are presented in Table III with the theoretical values for the acid chloride. TABLE III ELEMENTAL ANALYSIS OF THE ACID CHLORIDE OF TMA C, % H, % Analysis 51.35 1.75 30.19 16.71 Theoretical 51.33 1.44 30.39 16.84 0, % Cl, % It was also determined that the acid chloride could be readily purified by dissolving the sample in anhydrous ether at room temperature followed by cooling in acetone-dry ice, m.p. 67° . Sublimation of the crude acid chloride at 50° and 1-2 mm. also led to the isolation of fine, white crystals, m.p. 670. REACTION OF THE ACID CHLORIDE AND ALCOHOLS The acid chloride of TMA should react readily with alcohols to form esters. As this reaction should yield primarily the ester anhydride, the reaction was -39investigated. Since the methyl ester anhydride of TMA (4-carbomethoxy phthalic anhydride) is a known compound, the emphasis of this study was placed on esterification with methanol. This ester was synthesized independently from TMA and ethereal diazomethane for. identification purposes. (See Appendix V for this preparation.) Preliminary Esterifications Several reactions were conducted between the acid chloride of TMA and methanol. Reaction of the acyl halide was indicated by the evolution of hydrogen chloride, but sirups were always obtained. anhydride ester were made. Numerous unsuccessful attempts to crystallize the It was not until the sirups had been standing for several weeks that sublimation was attempted. Sublimation, followed by low-temper- ature recrystallization from ether, led to the isolation of pure 4-carbomethoxy phthalic anhydride. Experimental details of the various reactions are presented in the following paragraphs. A single reaction with butanol-2 led to a sirup. Reaction 1 The acid chloride (5.6 g., 0.0266 mole) was dissolved in 10 ml. benzene at room temperature and filtered. Methanol (0.871 g., 0.0272 mole) was added, with immediate evolution of heat and of hydrogen chloride. The mixture was allowed to cool slowly without precipitation and was eventually concentrated to a sirup. Cooling and scratching failed to initiate crystallization. a precipitate was observed in the sirup. After about seven weeks, The sirup was diluted with 25 ml. benzene, which led to the isolation of three precipitates. The first crop (0.25 g.) was trimellitic acid, m.p. 217°d. and negative ferric hydroxamate test. product (0.2 g.) range (62-183° ). had m.p. 165-180 °. The third crop (0.19 g.) This was extracted with 1,2-dichloroethane. The second melted over a wide The recrystallized -40- portion (0.02 g.) softened at 176-178 ° and melted by 199-205° . a negative ferric hydroxamate test. An infrared spectrum indicated the sample was a monomethyl trimellitate (carboxyl absorption at 1710 cm. 1 ion at 1734 cm. 203.5-205.5 ° (1). concentration. 189 ° . ). This sample gave 1 and ester absorp- The monoester 1-methyl trimellitate is dimorphic, m.p. 177, The remainder of the dichloroethane extract was a sirup on The fraction insoluble in dichloroethane (0.11 g.) had m.p. 183- This material was insoluble in benzene and after recrystallization from dioxane had m.p. 180-186° . leading to a sirup. The remaining benzene solution was concentrated, again A small portion (0.01 g.) of the impure ester anhydride was obtained, however, on sublimation, m.p. 85-93 °. Additional reactions were conducted in different solvents, at different temperatures, and with varying rates of addition of methanol, but the usual result was a sirup. Details of these reactions with similar results are presented in Appendix VI. Reaction with Butanol The acid chloride (5.25 g., 0.025 mole) was dissolved in 10 ml. benzene and filtered. Butanol-2 (2.0 g., 0.027 mole) was added. The solution had to be warmed slightly to initiate the evolution of hydrogen chloride. to cool slowly and was eventually concentrated to a sirup. did not bring about crystallization. The mixture was allowed Cooling and scratching After seven weeks a precipitate was observed in the sirup, which was then diluted with benzene and filtered (0.09 g.), m.p. 199-205 ° . This product was insoluble in benzene, dichloroethane, carbon tetra- chloride, toluene, o-dichlorobenzene, and cyclohexane but soluble in dioxane and ether. On recrystallization from ether (0.06 g.) the sample had m.p. 187-191° and gave a positive ferric hydroxamate test. Infrared analysis indicated that the sample was mostly trimellitic acid with a small amount of a monobutyl ester present. -41- A small second product (0.02 g.) which separated from the benzene filtrate had m.p. 120-135 ° . Concentration again led to a sirup. and 0.8 mm. for fifteen hours was unsuccessful. A sublimation attempt at 80 ° No significant amount of sub- limate was recovered. Quantitative Study Although 4-carbomethoxy phthalic anhydride had been obtained from the above reactions of methanol and the acid chloride of TMA, the reactions were by no means quantitative. The sirups obtained from these reactions had been subjected to a certain amount of hydrolysis while standing for several weeks before sublimation was attempted. For this reason, an additional reaction was conducted to determine the maximum possible yield of 4-carbomethoxy phthalic anhydride which could be obtained from the acid chloride of TMA. The acid chloride was prepared in the usual manner and recrystallized from anhydrous ethyl ether, m.p. 66-68 °. Methanol (0.269 g., 0.0084 mole) was added dropwise to the finely divided acid chloride (1.75 g., 0.0083 mole). The reaction was accompanied by the evolution of heat and of hydrogen chloride and resulted in a sirup. This product was subjected to sublimation at 80° and 1 to 2 mm. After two weeks, 1.1663 g. crude 4-carbomethoxy phthalic anhydride were collected, m.p. 85-1010 (68.2% yield). Recrystallization from ether yielded 0.7 g., m.p. 94-101 °. A second recrystallization gave 0.4 g., m.p. 100-101°; mixed m.p. with 4-carbomethoxy-phthalic anhydride, 99-101 °. Analysis of 4-Carbomethoxy Phthalic Anhydride The methyl ester anhydride obtained from the reaction of methanol and the acid chloride of TMA usually had m.p. 100-101 ° . When mixed with a sample of the ester obtained from the reaction of TMA and ethereal diazomethane, the m.p. was 99-101 ° . -42- The molecular weight, determined by the freezing point depression of benzene, was 202 + 15 (theoretical: ization (see Fig. 1). 206.15). The infrared spectrum supported the character- No absorption due to carboxyl groups could be observed. There was carbonyl absorption (cyclic anhydride) at 1858 and 1779 cm. 1 as in TMA in addition to a strong ester absorption band at 1732 cm. analyses were made by the Geller Laboratories. -1 . Carbon and hydrogen Their results along with the oxygen content (by difference) are presented in Table IV with the theoretical values for 4-carbomethoxy phthalic anhydride. TABLE IV ELEMENTAL ANALYSIS OF 4-CARBOMETHOXY PHTHALIC ANHYDRIDE C, % H, % 0, % Analysis (1) 57.82 3.29 (2) 57.66 3.34 (Av.) 57.74 3.32 38.94 58.26 2.93 38.81 Theoretical ESTERIFICATION OF THE TRIANHYDRIDE WITH ALCOHOLS Throughout the course of this study, the trianhydride, or bis(trimellitic anhydride) anhydride, has been obtained as a product of several reactions. The trianhydride was produced primarily as a decomposition product of unsymmetrical anhydrides. Since this compound contains two cyclic anhydride groups, as in TMA, and one linear anhydride group, as in benzoic anhydride, it seemed of interest to conduct esterifications. PRELIMINARY REACTIONS The first esterification reaction consisted of refluxing the trianhydride in acetone with sec-butyl alcohol. Reaction conditions apparently were not strong -43- enough, however, as the trianhydride was recovered. Subsequent reactions were carried out with methanol, followed by successful identification of TMA and 4carbomethoxy phthalic anhydride in the reaction products. Butanol The trianhydride (8.05 g., 0.022 mole) was dissolved in 150 ml. dry acetone and refluxed eight hours with 1.63 g. butanol-2 (0.022 mole). The initial crop of fine white crystals which separated on cooling (2.6 g.) had m.p. 218-227° and an anhydride equivalent of 123 (theoretical for the trianhydride: 122.08). After three recrystallizations from acetone and o-dichlorobenzene, m.p. 220-225 ° , the product had an anhydride equivalent of 127. ably the same product, m.p. 221-226 ° . The second crop (0.73 g.) was prob- Attempts to recrystallize the third crop (1.95 g.), m.p. 140-215° , and the fourth crop (0.32 g.), m.p. 131-198°, from methyl ethyl ketone led to partially hydrolyzed products which gave weak ferric hydroxamate tests and decomposed on heating. Methanol The trianhydride was prepared by refluxing TMA (10 g., 0.0522 mole) and TFAA (13.5 g., 0.0642 mole) in 25 ml. acetone for five hours. On cooling, the trianhydride was filtered, washed with cold benzene, and dried, m.p. 214-222 ° and yield: 4.37 g. (45.8%). Some yellow coloration remained; this was removed with a cold carbon tetrachloride extract and a hot benzene extract, m.p. 216-222° . The trianhydride (3.65 g., 0.01 mole) was dissolved in 150 ml. hot 1,2-dichloroethane. After adding 0.32 g. (0.01 mole) methanol, the solution was refluxed sixteen hours. On cooling to -30°, 0.4 g. TMA separated, m.p. 162-166° . tration to 100 ml. led to the isolation of additional TMA, m.p. 161-167° . solution was eventually concentrated to a sirup. ConcenThe Various crystallization techniques -44- and extractions were attempted on this sirup leading to the isolation of several subcrops, all of which were mixtures. TMA was the only product identified. A second reaction was carried out with methanol. The trianhydride was pre- pared by the reaction of TMA and TFAA in acetone at room temperature. was filtered, washed with benzene, and dried, m.p. 222-226 ° . The product The trianhydride (2.9 g., 0.008 mole) was dissolved in hot 1,2-dichloroethane and 0.253 g. (0.008 mole) methanol were added dropwise. The solution was refluxed 24 hours and then vacuum concentrated to dryness at room temperature. The product was composed of both a solid and a sirup, and was extracted with cold benzene. The insoluble material (1.66 g.) had a m.p. of 108-145° and was again extracted with cold benzene followed by hot benzene. 162-165° . The recovered insoluble material was TMA, m.p. A small crop (0.08 g.) which recrystallized from the hot benzene had a m.p. of 108-145° . The remaining extract was a sirup on concentration. The first crop to separate from the cold benzene extract of the main product had a m.p. of 110-130° and a yield of 0.05 g. zene (0.02 g.). until 125 ° . This was recrystallized from ben- Most of the sample melted at 90-95 ° but the melt was not clear Some methyl ester anhydride was probably present in this sample. second crop from benzene (0.1 g.) had a m.p. of 180-205°d. The The remaining benzene extract was a sirup on concentration. After about seven weeks, a precipitate was observed in the sirup. dilution with 5 ml. ether, 0.01 g. were filtered off, m.p. 87-94 ° . After On recrystal- lization from benzene (0.005 g.), the m.p. was 89-93 ° (impure methyl ester anhydride). out. After evaporation of the ether, a sublimation attempt at 850 was carried The sublimate collected (0.03 g.) had a m.p. of 91-100° . On recrystalliza- tion from ether (0.01 g.) the m.p. was 100-101° (4-carbomethoxy phthalic anhydride). Reported m.p. values in the literature range from 94-105° (1-2). Additional -454-carbomethoxy phthalic anhydride was obtained by redissolving the product in ether and cooling to -80° for 24 hours. having a m.p. of 87-98°. This led to the precipitation of 0.08 g. Recrystallization from ether gave the anhydride ester (0.04 g.), m.p. 101-103°. QUANTITATIVE STUDY As in the case of the preliminary esterifications of the acid chloride of TMA, the above reaction of the trianhydride and methanol could not be considered as quantitative, since the resulting sirup must have been.subjected to a certain degree of hydrolysis. For this reason an additional reaction was conducted and immediately followed by sublimation. The trianhydride (0.89 g., 0.0024 mole) was dissolved in 50 ml. 1,2-dichloroethane. After the addition of 0.0792 g. methanol (0.0025 mole) the solution was refluxed eighteen hours. After concentrating the solution to dryness, the product (0.79 g.) had a m.p. of 100-170°. After subjecting this product to sublimation at 80° and 1 to 2 mm. for five days, 0.1 g. crude 4-carbomethoxy phthalic anhydride was obtained, m.p. 88-98° (yield: 20.2%). After low-temperature recrystallization from ether, 0.06 g. were recovered, m.p. 99-100 ° and mixed m.p. 99-101° . -46- DISCUSSION OF RESULTS AND CONCLUSIONS THE TRIMELLITIC ANHYDRIDE-TRIFLUOROACETIC ANHYDRIDE REACTION SYSTEM THE UNSYMMETRICAL ANHYDRIDE OF TMA AND TFAA The reaction of TMA and TFAA results in the formation of the unsymmetrical anhydride, which is actually a dianhydride consisting of a cyclic and a linear anhydride: 0 (CF CO)0O + HOO 0 _ -CO CFcOC 3 \O c \\ + CFCOOH. (25) (23) o The cyclic anhydride group is quite stable in this system. In fact, any tri- mellitic acid impurity would be converted into TMA in a manner analogous to the preparation of phthalic anhydride by treating phthalic acid with acetic anhydride: 0 C -COOH (CFCO) 2 0 + /0 HOOC OOH HOOC- + 2C2 CF COOH (24) C:O There can be no doubt that the product is an unsymmetrical anhydride and not merely an equimolar mixture of the simple anhydrides due to the melting point, which is much lower than that of TMA or its anhydride, and since the anhydride can be recrystallized from benzene. A mixture of anhydrides would also be unlikely because of the high volatility of TFAA. The values obtained for neutralization equivalent, anhydride equivalent, and fluorine content were consistent with the identification, as was the infrared spectrum of the purified product. be consistent for a polyanhydride such as: These analyses would also -47- 0 0 II 0 0- II -COOCOCF CF C-0-C which would require the opening of the cyclic anhydride ring, the stability of which has already been mentioned. However, this alternate structure was shown The molecular weight to be impossible by the determination of molecular weight. was obtained from the freezing point depression of benzene, a solvent in which the unsymmetrical anhydride is fairly stable. The unsymmetrical anhydride of TMA and TFAA readily undergoes partial hydrolysis, leading to the formation of TMA and trifluoroacetic acid: 4 °F1 ° CF3C-O- X CFCOOH S0 + H2 0 C^ HOOC C (25) o If an attempt is made to recrystallize this anhydride from a solvent which contains a small amount of water, hydrolysis will occur, as it must have when a sample was dissolved in carbon tetrachloride. It has been stated previously (17) that carboxylic trifluoroacetic anhydrides hydrolyze completely by standing in contact with moist air for even a few seconds. spectra. This would explain the infrared Moisture adsorbed from the air by potassium bromide led to a spectrum which was essentially the same as the known spectrum of TMA. When nujol was employed instead of potassium bromide there was less chance for hydrolysis to occur and the spectrum suggested the presence of the four carbonyl bands of a dianhydride. However, there was still slight carboxyl absorption, indicating that a small degree of hydrolysis had taken place. With a compound of the type RCOOCOR1 , the rate of hydrolysis would be strongly dependent on the role played by the alkyl group in decreasing the availability of electrons at the reaction site. If the hydrogen atoms of the alkyl group are re- placed by fluorine atoms, as in the trifluoroacetyl group, the R group is a very powerful electron-withdrawing group due to the strong inductive effect of the highly electronegative fluorine. Electron-withdrawing substituents increase the tendency for a water molecule to combine with the carbonyl carbon atom. The rate of hydrolysis is also increased by increasing differences in sizes of the two R groups. Thus, the highly reactive nature of the unsymmetrical anhydride of TMA and TFAA toward water would be expected. The unsymmetrical anhydride of TMA and TFAA disproportionates very easily: 3 ° °I 0 2 CF3 OC C / 2 00 = (CFC0) 2 0 + °\ O 0C °II I0 COC 2 (26) This disproportionation occurs under the influence of heat or certain solvents. When the anhydride is heated to its melting point, disproportionation takes place immediately leading to the evolution of TFAA and the solidification of bis(trimellitic anhydride) anhydride. Other unsymmetrical anhydrides in which the two alkyl or aryl groups differed considerably in size showed a marked tendency to disproportionate into the simple anhydrides even on standing at room temperature (28). This factor was also found to be in effect with the unsymmetrical anhydride of TMA and TFAA; it was found to be necessary to analyze this anhydride immediately -49- after preparation and purification. was submitted for elemental analysis. The instability was illustrated when a sample The carbon and hydrogen values obtained were intermediate to the calculated values for TMA and bis(trimellitic anhydride) anhydride, indicating that both disproportionation and hydrolysis had probably taken place. Although the unsymmetrical anhydride can be recrystallized from a nonpolar solvent, such as benzene, when it is dissolved in a more polar solvent, such as acetone, nitroethane, or ether, disproportionation takes place. The equilibrium of Reaction (26) must be strongly influenced by the nature of the solvent. The more polar solvents must shift the equilibrium to the right resulting in a preponderance of the simple anhydrides. This would agree with the results of Ralston and Reck (42) who could not recrystallize unsymmetrical anhydrides from acetone or acetonitrile, but could recrystallize from petroleum ether, even though the anhydrides were quite soluble in all three solvents. It has been demonstrated that perfluoro fatty acids form addition compounds with some ethers, tertiary amines, nitrites, and ketones (82). For example, tri- fluoroacetic acid formed an addition compound with ethyl ether while heptafluorobutyric acid formed addition compounds with acetone and acetonitrile. If TFAA could be complexed by ether, such as by intermolecular hydrogen bonding between fluorine atoms and the hydrogen atoms of ether, this might be expected to shift the equilibrium of Reaction (26) to the right, leading to the crystallization of bis(trimellitic anhydride) anhydride. position of the equilibrium. Solubility properties might also affect the The unsymmetrical anhydride readily dissolved in cold ether, and crystallization of bis(trimellitic anhydride) anhydride commenced within a few minutes. -50ESTERIFICATION OF THE TMA-TFAA SYSTEM One series of esterification reactions of the TMA-TFAA system was quite different from any of the other reactions. Reactions conducted in an acetone medium led to the isolation of bis(trimellitic anhydride) anhydride. Reactions carried out in acetone must be strongly influenced by the position of the equilibrium of Reaction (26). This would be analogous to the attempt to recrystal- lize the unsymmetrical anhydride from ether. Indeed, when no alcohol was added to the solution of TMA and TFAA in acetone, bis(trimellitic anhydride) anhydride, the trianhydride, crystallized out in 50% yield. Assuming that the equilibrium is displaced to the right a certain degree, when an alcohol is added, it could react with the trianhydride, TFAA, or the unsymmetrical anhydride. The latter two compounds would be expected to be the most reactive of the three. When a slightly less than equimolar quantity of phenol or o-cresol was added to the esterification mixture, the trianhydride was the first compound to separate. However, the trianhydride was never recovered in pure form in yields greater than 15%. This would indicate that in addition to the second disproportionation product (TFAA) reacting with alcohol, the unsymmetrical anhydride was also reacting. If the unsymmetrical anhydride reacted to give mainly the trifluoroacetate ester, TMA should also be formed. TMA was recovered from these esterification reactions in acetone in yields up to 50%. To show that the trifluoroacetate ester was actually being formed, n-butyl trifluoroacetate was recovered from one reaction of TMA, TFAA, and n-butyl alcohol in acetone. For the general esterification of the TMA-TFAA system, where the unsymmetrical anhydride would be expected to predominate, there are two possible reaction mechanisms. The first type would be the ionic mechanism which is usually encoun- tered for reactions of acyl trifluoroacetates. In this case the unsymmetrical -51- anhydride would ionize a small extent into a trifluoroacetate anion and a trimellityl cation. (The alternative mode of ionization into a trifluoroacetyl cation and a trimellitate anion would be quite unlikely due to the strong electronegative character of fluorine.) The reactive acylium ion would then be the primary acylat- ing species, leading to a trimellitate ester anhydride and trifluoroacetic acid: 0 II /°0 0 CF3C-O-C (CF3COO) + 1 0 +C-I (27) C O ROH 1 CF COOH+ (28) C/ O ROOC In the second type of reaction mechanism, the bimolecular displacement reaction suggested by Tedder (56), the alcohol would react directly with the molecular form of the unsymmetrical anhydride. This mechanism can be used to predict the results for the esterification of most unsymmetrical anhydrides. Since nucleophilic attack occurs preferentially at the carbonyl group which is most deficient in electrons, this mechanism would favor the formation of the ester of the stronger acid. For the esterification of the unsymmetrical anhydride of TMA and TFAA, this mechanism would predict, therefore, that the trifluoroacetate ester and TMA would be formed: riT.i r0/-in)( r\ I[ n \ .\ /O + CF3COOR + ROH ROH-->CF 3 COOR ++ 1f1 n /0 HOOC I /0 (29) (29) -52- In most of the esterification reactions, approximately equimolar quantities of TMA and alcohol were employed, along with a slightly greater than equimolar quantity of TFAA. This procedure was followed to minimize esterification with the cyclic anhydride ring of TMA and with excess TFAA. Under conditions unfavorable for the ionization of the unsymmetrical anhydride, the bimolecular reaction resulting in TMA and the trifluoroacetate ester would be expected to be in effect. out in the chlorinated solvents. This was demonstrated by the reactions carried A reaction in carbon tetrachloride with phenol and reactions with sec-butyl alcohol in o-dichlorobenzene and 1,2-dichloroethane led to the recovery of TMA in yields of better than 80%. The alcohol must have reacted with the molecular form of the unsymmetrical anhydride in these cases. In the former reaction a minor crop was obtained which could have contained the phenyl trimellitate anhydride, but insufficient material was obtained for positive identification. The reaction of an equimolar portion of alcohol with the solid unsymmetrical anhydride also primarily follows the bimolecular reaction. with phenol led to the recovery of TMA in 84% yield. The esterification When the unsymmetrical anhy- dride was reacted with n-butyl alcohol, crude TMA in about 80% yield was obtained. The distillate from this reaction amounted to 69% yield, based on n-butyl trifluoroacetate. The esterification reaction of methanol with the solid unsymmetri- cal anhydride led to the isolation of a product which was mostly TMA (87% yield). It has been shown that 4-carbomethoxy phthalic anhydride can be removed from mixtures by sublimation. In this case the sublimate softened slightly at about the melting point of the ester anhydride, but was apparently composed primarily of TMA. Very little trimellitate ester could have been formed in this reaction, and the trifluoroacetate ester must predominate. Only the sharpest fraction of methyl -53trifluoroacetate was retained from the distillate of this reaction (26.2% yield). The actual yield must have been considerably higher, but a large part of the ester was lost due to its high volatility. A reaction of the unsymmetrical anhydride in ether with n-butyl alcohol, led to TMA and n-butyl trifluoroacetate. It would be of interest at this point to compare the results of this study with those obtained by Bourne, et al. (72) for the esterification of the unsymmetrical anhydride of TFAA and benzoic acid, which might be expected to be similar to the unsymmetrical anhydride of TMA and TFAA. Benzoyl trifluoroacetate was a more effective trifluoroacetylating agent than an unsymmetrical anhydride like acetyl trifluoroacetate, probably due to resonance stabilization of the benzoylium cation, but some benzoate esters were obtained. With n-butyl alcohol, Bourne obtained 6% benzoate and 93% trifluoroacetate; in a nonionizing solvent (carbon tetrachloride) he obtained 97% trifluoroacetate. When the reaction was conducted in an equimolar portion of trifluoroacetic acid (1.3 moles), the result was 8% benzoate and 92% trifluoroacetate. The result under the same conditions for esterification with the secondary alcohol was the same, but reaction with phenol led to 88% benzoate and 11% trifluoroacetate. (Since phenol is appreciably acidic, the electrons of the oxygen atom should be relatively less readily available for reaction with the weaker electrophilic reagent than with the benzoylium ion, and attack by the molecular form of the unsymmetrical anhydride should be slowed down.) Reaction with ethanol led to 13% benzoate and 81% trifluoroacetate. When a large quantity (10 moles) of trifluoroacetic acid was employed as a solvent, reaction with n-butyl alcohol led to 38% benzoate and 59% trifluoroacetate. Sub- stitution of the secondary alcohol in this reaction system led to 57% benzoate and 40% trifluoroacetate. The decreasing proportion of trifluoroacetate obtained from the secondary alcohol is explained by the greater difficulty of approach of the molecular form of the anhydride to the oxygen atom of the alcohol. -54In this study, no trimellitate ester anhydride was isolated when alcohol was added to the unsymmetrical anhydride alone, or to the anhydride in nonionizing solvents. Trifluoroacetate esters and TMA were obtained. A reaction with approxi- mately equimolar portions of methanol and the unsymmetrical anhydride in ten moles of trifluoroacetic acid was followed by sublimation of the dried product. The results indicated that very little 4-carbomethoxy phthalic anhydride could have been formed. If the reaction were similar to that of benzoyl trifluoroacetate, a greater amount of trimellitate ester would be formed in the reaction with a secondary alcohol. Four reactions were conducted between equimolar quantities of the unsymmetrical anhydride and sec-butyl alcohol in trifluoroacetic acid (three to ten moles). Reactions were carried out at room temperature and at reflux. In all cases, TMA was obtained in high yields and no trimellitate ester could be identified positively. had the same result. Reaction with a longer chain secondary alcohol (capryl) Equimolar quantities of the alcohol and the unsymmetrical anhydride in ten moles of trifluoroacetic acid led to the almost immediate crystallization of 72.4% TMA. The reaction of phenol and benzoyl trifluoroacetate led to a high yield of phenyl benzoate, but with the unsymmetrical anhydride of TMA and TFAA, TMA was recovered in large quantities. A very small amount (0.02 g.) of a monophenyl trimellitate was obtained but was not completely characterized. Various recrystallization and extraction techniques were employed in the above reactions, but TMA was always obtained. The reactions of alcohols with the unsymmetrical anhydride of TFAA and TMA appear to be quite different from the reactions of alcohols with acetyl and benzoyl trifluoroacetate. All of the reactions conducted led to TMA and tri- fluoroacetates almost exclusively. molecular reaction of the SV2 type. In all cases the reaction must follow the biIf the reaction followed the usual ionization -55mechanism of the trifluoroacetyl unsymmetrical anhydrides, the trimellitate esters would have been obtained. Even under conditions favoring the ionization mechanism, such as the reactions carried out in excess trifluoroacetic acid, and with alcoholic compounds usually favoring this mechanism, such as secondary alcohols and The mechanism postulates phenols, the trimellitate esters could not be obtained. a small degree of ionization into the trifluoroacetate and acylium ions. A partial explanation why this mechanism is not favored, is that resonance stabilization of any acylium cations formed leads to accompanying lowering of the density of the positive charge on the carbonyl group: o II\t +C -C "^ ) I 'oI C O o > J C-C C ° o ZO -O II C-C 0+ °oC o - ] Ie> 11 C -> (30) This may be a simplification due to the influence of the anhydride group, but would have the effect of slowing down the reaction of the cation with hydroxyl groups and allow the reaction with the molecular form of the unsymmetrical anhydride to become more important. This explanation is not completely satisfactory, since similar resonance structures can be proposed for the benzoylium cation. It does partly explain, along with steric effects, why greater proportions of trifluoroacetates are obtained from the unsymmetrical anhydrides with TMA and with benzoic acid than are obtained from acetyl trifluoroacetate. However, since resonance structures for the benzoylium ion would be more highly favored than those of the trimellitylium ion, on this basis alone higher proportions of trimellitate esters than benzoate would be expected from their corresponding acyl trifluoroacetates. -56It would appear that very little ionization actually takes place. It has been stated (83) that benzyl carbonium ions normally are formed with ease, since they are stabilized by resonance, provided that electron-withdrawing groups are not conjugated with the system. For example, when a carbonyl group is conjugated with the system, resonance operates to oppose the separation of the ions; under these conditions carbonium ion formation is unlikely. The interpretation of the effects of substituents on a reacting molecule are divided into inductive (or electrostatic) effects, resonance (or conjugation) effects, and steric effects (84). The Hammett substituent constant (a) represents the ability of the substituent group to attract or repel electrons by a combination of its inductive and resonance effects. The constant is not applicable to ortho substitution due to steric effects, but it is meta and para substitution in effect in this instance. The acid-strengthening substituents, those with positive a's, are classed as electron-withdrawing agents (85). There are no values of a for anhydride groups in the literature, but these meta and para carbonyls might be expected to be similar to meta and para methyl ester groups. Values of a for meta and para -COOCH groups are both strongly positive, although not as much so as the' values for -CF3 or -N02 groups. Both carbonyl groups con- tain an electronegative atom doubly bonded to a more positive atom that is in turn singly bonded to the rest of the molecule. The carbonyl oxygens can withdraw electron density from the neighboring carbon atoms leaving them with partial positive charges. This inductive effect arising from the action of dipoles results in the atoms nearest the position of substitution being subjected to the greatest induced polarization. However, it appears that inductive effects are transmitted to the reaction center almost as efficiently from the para as from the meta position (85). Carbonyl groups, which are associated with negative inductive effects, -57also withdraw pi electron density from conjugated systems. This disturbance of pi electron density at one atom in a conjugated system is not diminished by distance. The best indications of the resonance effect are exhibited with para substituents. These effects due to the meta and para carbonyls would both act to withdraw electron density. They would not be expected to be as great as the effect of the -CF3 group of the other half of the unsymmetrical anhydride, but would act in opposition to its electron-withdrawing action. The over-all effect might be a diminution of ionization, resulting in the pronounced reaction of the alcohol with the molecular form of the unsymmetrical anhydride. Under these circumstances, the formation of TMA and trifluoroacetates would be expected due to the action of the more electronegative -CF3 group. Three reactions were conducted in which a greater than equimolar quantity of alcohol was added to the unsymmetrical anhydride. These reactions all re- sulted in the opening of the cyclic anhydride ring with the consequent formation of 1- and 2-alkyl trimellitates. The reaction of excess methanol with the solid unsymmetrical anhydride led to a mixture of 1- and 2-methyl trimellitates. The reaction in chloroform with excess n-butyl alcohol led to a monobutyl trimellitate. A reaction of one mole of the anhydride with two moles sec-butyl alcohol in trifluoroacetic acid also led to the opening of the ring and formation of monobutyl trimellitates. THE UNSYMMETRICAL ANHYDRIDE OF TRIMELLITIC AND ACETIC ANHYDRIDES The unsymmetrical anhydride of TMA and acetic anhydride may be obtained, as indicated in Reaction (7), by refluxing a solution of the two anhydrides. The unsymmetrical anhydride does not separate out on cooling to room temperature, as in the case of the trifluoroacetyl derivative. It is necessary to distill off -58- some of the excess solvent and follow by cooling to obtain the crude derivative. The anhydride cannot be obtained by continued distillation to remove acetic acid and acetic anhydride, as this would lead to disproportionation, as shown in Reaction (8). The unsymmetrical anhydride can be purified either by extraction of the impurities with ether or by extraction of the unsymmetrical anhydride with benzene followed by recrystallization from the same solvent. This product could not be merely a mixture of the two anhydrides since it has a distinct melting point and it can be recrystallized. Analytical values were consistent with the identi- fication, as was the infrared spectrum. The determination of molecular weight eliminated the possibility of a polyanhydride. The esterification of this unsymmetrical anhydride was only briefly examined. Three reactions were carried out with methanol to determine the possibility of obtaining 4-carbomethoxy phthalic anhydride from this unsymmetrical anhydride. The reactions were all followed by sublimation, but the ester anhydride was not obtained in pure form. Again this reaction might be similar to that of acetic benzoic anhydride which leads predominantly to acetate esters (48). Without the influence of the highly electronegative -CF3 group, the ionic mechanism would not be favored in the case of acetic benzoic anhydride. The explanation of Bailey and Chang (48), due to the structure shown below, was that resonance in the benzoyl + )=c-O-C \-A /I O- -CH3 -59group would disperse the positive charge into the benzene ring making this carbonyl group less susceptible to nucleophilic attack than the acetyl carbonyl group, which would be polarized by the inductive effect of the methyl group. In the case of the acetic trimellitic anhydride, electronic effects, due to the influence of the carbonyls of the cyclic anhydride, would be.expected to make the trimellityl carbonyl carbon more positive, and would render reaction at this site more favorable than at the acetyl carbon atom. Since 4-carbomethoxy phthalic anhydride was The not obtained it would seem that this reaction was governed by steric effects. much greater size of the phenyl group in comparison to the methyl group would favor the formation of acetate esters and TMA. THE ACID CHLORIDE OF TRIMELLITIC ANHYDRIDE The two anhydride derivatives of TMA discussed above react with alcohols to return TMA. It was desirable to obtain an anhydride derivative which could react The with alcohols to yield high percentages of the trimellitate ester anhydrides. acid chloride of TMA appeared to be the most promising derivative. Refluxing a mixture of TMA and excess thionyl chloride until solution is obtained leads to the formation of the acid chloride: 0 + SOC1 C =C- ^ COOH -COOH -2C > O + S C -r + HC1 (31) -COC1 On cooling the solution to room temperature, a small amount of bis(trimellitic anhydride) anhydride crystallizes, probably formed by the reaction of the acid chloride with TMA: -60- <+ 3 + XCOCd ) --- ^+HC1 + + HOOCHCOC 0 0 Ci Il 0\ II c (32) > Vacuum concentration of the filtrate to dryness leads to high yields of the acid chloride. The acid chloride of TMA may be obtained quite easily in pure form by recrystallization from either benzene or ether. obtained by sublimation. Fine, white crystals may also be Elemental analysis, neutralization equivalent, molecu- lar weight, and infrared analysis were all consistent with the proposed structure. The acid chloride portion of the molecule should be more reactive than the cyclic anhydride portion. When methanol was added to the solid acid chloride or to a benzene or 1,2-dichloroethane solution, an immediate exothermal reaction was indicated with evolution of HC1: oc0 00 *oC + CIHOH -> <C-- COCl ° C OC Concentration of the products led to a sirup. + HCl (33). COOCH3 Varying the reaction conditions and the rate of addition of methanol had no effect, and crystallization could not be induced. The reaction with butanol, accompanied by the evolution of hydrogen chloride, also led to a sirup. Since hydrogen chloride was evolved, it was known -61- that the acid chloride was reacting. However, since crystallization could not be induced it seemed possible that this was not the sole reaction. There could be a small degree of esterification of the cyclic anhydride ring; this reaction could be catalyzed by the hydrogen chloride evolved. It still seemed probable that the main reaction was the esterification of the carboxylic acid chloride. It has been shown (86) for acylation of substituted benzoyl chlorides, that reactivity runs parallel with the depletion of electrons at the carbonyl carbon atom. It might be expected that the anhydride group should increase the reactivity of the acid chloride. Gould (84) has stated that alcoholysis of acyl chlorides is typical of the bimolecular nucleophilic substitution reactions, and that the reaction is accelerated by the substitution of electron-withdrawing groups, such as -NO2, in the benzene ring. It was discovered that 4-carbomethoxy phthalic anhydride could be removed from the sirupy product by sublimation. By this procedure, the crude ester was obtained in 68.2% yield, indicating that esterification of the acyl chloride was the main reaction. This ester was purified by recrystallization from ether. Infrared analysis, elemental analysis, melting point, and the molecular weight determination were all consistent with the identification. BIS(TRIMELLITIC ANHYDRIDE) ANHYDRIDE The simple anhydride of TMAwas obtained by several preparations during this work, primarily through disproportionation of an unsymmetrical anhydride. Heating of the unsymmetrical anhydride of TMA and TFAA or solution in certain solvents led to disproportionation and the recovery of bis(trimellitic anhydride) anhydride. This trianhydride has been purified by recrystallization from various solvents. The determination of melting point, neutralization equivalent, anhydride equivalent, infrared analysis, and elemental analysis were all consistent with the identification. -62- Once again, the possibility of a polyanhydride was eliminated by the determination of molecular weight. A few esterification reactions of the trianhydride were conducted. It might be expected that the linear anhydride group could be slightly more reactive than the cyclic anhydride groups, so that TMA and a 4-carboalkoxy phthalic anhydride would be obtained: °o o0 .o0 I II -C O-C + ROH -\ ---- > C ZC 0> P + O/C ,COOH ROOC (34) -c Steric factors might slow down this reaction, however, leading to some esterification of the cyclic anhydride groups. as the unsymmetrical anhydrides. This simple anhydride is not as reactive When dissolved in acetone and refluxed with sec- butyl alcohol, a considerable portion of the unchanged trianhydride crystallized out on subsequent cooling. Reaction was obtained, though, with methanol. As in the case of reaction with the acid chloride, concentration of the reaction mixture led to a sirupy product. lated from the product. Both TMA and 4-carbomethoxy phthalic anhydride were isoThis reaction is not as specific as the reaction of the acid chloride, however, as sublimation resulted in the recovery of only 20.2% of the theoretical yield of 4-carbomethoxy phthalic anhydride. SUMMARY OF RESULTS Five anhydride derivatives of TMA have been obtained. The trifluoroacetyl and acetyl unsymmetrical anhydrides, which are new compounds, were prepared, -63purified, and characterized. These unsymmetrical anhydrides are difficult to pur- ify since they readily hydrolyze and disproportionate. They react with alcohols leading predominantly to TMA and the trifluoroacetate or acetate ester. The acid chloride. of TMA, a previously unreported compound, can be obtained through the reaction of TMA with thionyl chloride. purified and characterized. This acid chloride has been Reaction with methanol leads to a sirup. Sublimation, however, removes the desired 4-carbomethoxy phthalic anhydride in high yield. This ester anhydride has also been purified, and characterized more completely than in previous reports of the compound. The acid chloride would be the most promising anhydride derivative for further work in this area, such as in the preparation of the 4-carboalkoxy-phthalic anhydride series, simple and mixed diesters, and ultimately, cross-linked esters. The simple anhydride of TMA, bis(trimellitic anhydride) anhydride, has been prepared by the disproportionation reaction of the unsymmetrical anhydride of TMA and TFAA and from the reaction of TMA and thionyl chloride. purified and characterized. was initiated. This derivative was A report of this compound appeared after this study Reaction with methanol led to TMA and 4-carbomethoxy phthalic anhydride, but the reaction does not appear to be as specific as the reaction of the acid chloride. -64ACKNOWLEDGMENTS The author wishes to gratefully acknowledge the assistance and suggestions of his thesis advisory committee: Kyle Ward, Jr., J. W. Green, and B. L. Browning. The author also wishes to express appreciation to Donald Johnson for his assistance and suggestions and to Lowell Sell for preparing the infrared spectra. -65LITERATURE CITED 1. Wegscheider, R., Perndanner, H. F., and Auspitzer, O., Monatsh. Chem. 31:1253-1301(1910). 2. Duff, S. R., Erdtman, H., and Harvey, W. E., Acta Chem. Scand. 8:1073-82 (1954). Dec. 1959. 10, 3. Amoco Chemicals Corp. Data Sheet, Ref. No. 2571-6-59, 4. Schulz, G., and Howard, H. C., J. Am. Chem. Soc. 68:991-4(1946). 5. Gonzalez-Sanchez, F., Tetrahedron 1:231-7(1957). 6. Bannister, B., and Elsner, B. B., J. Chem. Soc. 1951:1061-8(1951). 7. Feist, F., Ann. 496:99-122(1932). 8. Amoco Chemicals Corp. Trimellitic anhydride. Chicago, The Corp., 1958. 32 p. 9. Personal communication, 1961. Schubert, A. F., Amoco Chemicals Corp. 10. Barker, R. G. The esterification of cellulose with trimellitic anhydride. Unpublished work, The Institute of Paper Chemistry, 1960. 11. Malm, C. J., Svensk Kem. Tid. 73:523-30(1961). 12. Knobloch, J. 0., and Liao, H. P., U. S. patent 2,911,416(Nov. 3, 1959). 13. (a) (b) (c) 14. (a) Chiozza, L., Ann. 84:106-9(1852). (b) Chiozza, L., Ann. 85:225-33(1853). (c) Chiozza, L., Ann. 91:102-13(1854). 15. Kraut, K., and Hartmann, F., Ann. 133:99-108(1865). 16. Vaughn, J. R., and Osato, R. L., J. Am. Chem. Soc. 73:5553-5(1951). 17. Ferris, A. F., and Emmons, W. D., J. Am. Chem. Soc. 75:232-3(1953). 18. (a) Autenrieth, W., Chem. Ber. 20:3187-91(1887). (b) Autenrieth, W., Chem. Ber. 34:168-87(1901). Autenrieth, W., and Thomae, G., Chem. Ber. 57:423-37(1924). (c) 19. Askenasy, 20. Bourne, E. J., Gerhardt, C., Ann. 82:127-32(1852). Gerhardt, C., Ann. 83:112-16(1852). Gerhardt, C., Ann. 87:57-84(1853). P., and Meyer, V., Stacey, M., 1954:2006-12(1954). Chem. Ber. 26:1354-70(1893). Tatlow, J. C., and Worrall, R., J. Chem. Soc. -6621. Bucher, J. E., and Slade, W. C., J. Am. 22. Conix, A., Makromol. Chem. 24:76-8(1957). 23. Barnett, E. deBarry. Mechanism of organic chemical reactions. New York, Interscience, 1956. 24. Sonntag, N. O. V., Trowbridge, J. R., and Krems, I. J., Soc. 31:151-7(1954). 25. Wedekind, E., Chem. Ber. 34:2070-7(1901). 26. Wieland, T., and Stimming, D., Ann. 579:97-106(1953). 27. (a) (b) (c) (d) 28. Dunbar, R. E., and Garven, F. C., Proc. N. Dakota Acad. Sci. 3:24-7(1949). 29. Diels, 0., 30. Staudinger, H., Ann. 356:51-123(1907). 31. Adams, R., Wirth, W. V., and French, H. E., J. Am. Chem. Soc. 40:424-31(1918). 32. Cattelain, E., Bull. soc. chim. France 41:352-6(1927). 33. (a) (b) 34. Stetter, H., Marx-Moll, L., 35. Stevens, W. H., and Holland, D. A., Science 112:718-19(1950). 36. Ott, E. 1943. 37. Ott,.E., Langenohl, A., and Zerweck, W., Chem. Ber. 70:2360-2(1937). 38. Kirsanov, A. V., and Abrazhanova, E. A., Sbornik Statei Obshchei Khim. 2:865-8 (1953); C.A. 49:6820. 39. Eliel, E. L., 40. Kilpatrick, M., and Kilpatrick, M. L., 41. Zeavin, J. M., and Fisher, A. M., J. Am. Chem. Soc. 54:3738-42(1932). 42. Ralston, A. W., and Reck, R. A., J. Org. Chem. 11:624-6(1946). Hurd, Hurd, Hurd, Hurd, C. C. C. C. D., D., D., D., and and and and Chem. Soc. 31:1319-21(1909). J. Am. p. 172. Oil Chem. Dull, M. F., J. Am. Chem. Soc. 54:3427-31(1932). Thomas, C. L., J. Am. Chem. Soc. 55:275-83(1933). Williams, J. W., J. Am. Chem. Soc. 58:962-8(1936). Roe, A. S., J. Am. Chem. Soc. 61:3355-9(1939). and Lalin, L., Chem. Ber. 41:3426-34(1908). Naamlooze Vennootschap, British patent 12,042(May 23, 1913); C.A. 8:3618. Naamlooze Vennootschap, French patent 461,540(May 16, 1913); J. Soc. Chem. Ind. 33:219(1914). Organic syntheses. and Rutzen, H., Chem. Ber. 91:1775-81(1958). Vol. 2. p. 528. New York, John Wiley and Sons, and Burgstahler, A. W., J. Am. Chem. Soc. 71:2251-2(1949). J. Am. Chem. Soc. 52:1418-27(1930). -6743. Emmons, W. D., McCallum, K. S., and Ferris, A. F., J. Am. Chem. Soc. 75:6047-8 (1953). 44. Berliner, E., and Altschul, L. H., J. Am. 45. Gold, V., and Jefferson, E. G., J. Chem. Soc. 1953:1416-18. 46. Graves, G., U. S. patent 2,135,709(Nov. 47. Rollett,.A., and Scholz, F., Monatsh. 59:1-6(1932); 48. Bailey, P. S., and Chang, Y., J. Org. Chem. 27:1192-7(1962). 49. Polya, J. B., and Spotswood, T. M., J. Am. 50. Baroni, A., Marrano, G., and Modigliani, --. , Gazz. chim. Ital. 63:23-37(1933); C.A. 27:3447. 51. Polya, J. B., and Spotswood, T. M., Rec. Trav. Chim. 67:927-41(1948). 52. Kahn, R., Chem. Ber. 36:2535-8(1903). 53. Emery, A. R., and Gold, V. J., Chem. Soc. 1950:1455-60. 54. Emery, A. R., and Gold, V., J. Chem. Soc. 1950:1443-7. 55. Day, J. N. E., and Ingold, C. K., Trans. Faraday Soc. 37:686-707(1941). 56. Tedder, J..M., Chem. Revs. 55:787-827(1955). 57. Emery, A. R., and Gold, V., J. Chem. Soc. 1950:1447-55. 58. Treffers, H. P., and Hammett,.L. P., J. Am. Chem. Soc. 59:1708-12(1937). 59. Newman, M. S., J. Am. Chem. Soc. 63:2431-5(1941). 60. Stacey, M., Bourne, E. J., Chem. Soc. 74:4110-13(1952). 8, 1938); C.A. 33:1347. C.A. 26:1916. Chem. Soc. 71:2938(1949). Tatlow, J. C., and Tedder, J. M., Nature 164:705 (1949). 61. Bourne, E. J., 1949:2976-9. Stacey, M., Tatlow, J. C., and Tedder, J. M., J. Chem. Soc. 62. Morgan, P. W., Ind. Eng. Chem. 43, no. 63. Hamalainen, C., Wade, R. H., and Cruz, M. D., Textile Research J. 29:821-6 11:2575-7(1951). (1959). 64. Hamalainen, C., Wade, R. H., and Buras, E. M., Textile Research J. 27:168 (1957). 65. Ahlbrecht, A. H., and Codding, D. W., J. Am. Chem. Soc. 75:984(1953). 66. Morgan, P. W., J. Am. Chem. Soc. 73:860-1(1951). -68Tatlow, J. C., and Worrall, R., J. Chem. Soc. 1957:315-18. 67. Bourne, E. J., 68. Bourne, E. J., Randles, J. E. B., Stacey, M., Tatlow, J. C., and Tedder, J. M., J. Am. Chem. Soc. 76:3206-8(1954). 69. Randles, J. E. B., Tatlow, J. C., and Tedder, J. M., J. Chem. Soc. 1954:436-41. 70. Bourne, E. J., Randles, J. E. B., Tatlow, J. C., and Tedder, J. M., Nature 168:942-3(1951). 71. Reed, R., J. Am. 72. Bourne, E. J., Stacey, M., Tatlow, J. C., and Worrall, R., J. Chem. Soc. 1958:3268-82. 73. Bourne, E. J., 74. Filler, R., Fenner, J. V., Stokes, C. S., O'Brien, J. F., and Hauptschein, M., J. Am. Chem. Soc. 75:2693-5(1953). 75. Moffat, A., and Hunt, H., J. Am. Chem. Soc. 79:54-6(1957). 76. Benerito, R. R., Berni, R. J., 393-9(1960). 77. Burton, H., and Praill, P. F. G., Quart. Rev. 6:302-18(1952). 78. Hodes, W., U. S. patent 2,887,497(May 19, 1959). 79. Pasternak, T., Helv. Chim. Acta 23:1046-53(1940). 80. Larner, B. W., and Peters, A. T., J. Chem. Soc. 1952:680-6. 81. Bellamy, L. J. The infra-red spectra of complex molecules. New York, John Wiley, 1959. 82. Hauptschien, M., and Grosse, A. V., J. Am. Chem. Soc. 73:5139-41(1951). 83. Alexander, E. R. Principles of ionic organic reactions. Wiley, 1950. 318 p. 84. Gould, E. S. Mechanism and structure in organic chemistry. Henry Holt and Co., 1959. 790 P. 85. Hine, J. 552 p. 86. Williams, E. G., and Hinshelwood, C. N., J. Chem. Soc. 1934:1079-84. 87. Hogsett, J. N., Kacy, H. W., and Johnson, J. B., Anal. Chem. 25:1207-11(1953). 88. Johnson, J. B., and Funk, G. L., Anal. Chem. 27:1464-5(1955). Chem. Soc. 77:3403-5(1955). Tatlow, C. E. M., and Tatlow, J. C., J. Chem. Soc. 1950:1367-9. and Fagley, T. F., Textile Research J. 30: Physical organic chemistry. 2nd. ed. 2nd. ed. p. 128. New York, John New York, New York, McGraw-Hill, 1962. -6989. Steinbach, O. F., and King, C. V. Experiments in physical chemistry. York, American Book Co., 1950. 250 p. 90. Schoniger, W., Mikrochim. Acta 1955:123-9; 1956:869-76. 91. Brunisholz, G., and Michod, J., 92. Technological Service of the Arthur H. Thomas Co. 6470-E Thomas-Schoniger combustion flask. Philadelphia, 1958. 13 P. 93. Rogers, R. N., and Yasuda, S. K., Anal. Chem. 31:616-17(1959). 94. Johnson, C. A., and Leonard, M. A., Analyst 86:101-4(1961). 95. Belcher, R.,.Leonard, M. A., and West, T. S., J. Chem. Soc. 1959:3577-9. 96. Steyermark, A., Kaup, R. R.,.Petras, D. A.,.and Bass, E. A., Microchem. J. III:523-7(1959). 97. Senkowski, B. Z., Wollish, E. G., and Shafer, E. G. E., Anal. Chem. 31: New Helv. Chim. Acta 37:598-602(1954). 1574-6(1959). 98. Weygand, C. 99. Bhati, A., J. Org. Chem. 27:1183-6(1962). Organic preparations. p. 178. New York,.Interscience,.1945. -70- APPENDIX I DETERMINATION OF ANHYDRIDE EQUIVALENT The method employed for the determination of anhydride equivalents was based on the reaction of the anhydride with morpholine and titration of the excess reagent with hydrochloric acid (87-88). This method was chosen because of its reported success in the presence of acids and its determined applicability in the presence of esters. The required amount of redistilled morpholine was diluted with methanol to give a 0.1N solution. A 0.1N methanolic hydrochloric acid solution was stand- ardized against a sodium hydroxide solution which had been standardized previously with potassium acid phthalate. The indicator used was methyl yellow (p-dimethyl- aminoazobenzene) and methylene blue in methanol. The procedure consisted of adding an excess of the morpholine solution to the anhydride, allowing five minutes for reaction, followed by titration of the excess morpholine with the methanolic hydrochloric acid solution. It is also necessary to run a blank determina- tion. The mixed indicator solution suggested by Johnson and Funk goes through a wide color change which was not too important with the large sample sizes employed in their study, but with small samples this introduces a problem in determining the end point. This problem was eliminated, however, by following the titration potentiometrically. It was also found that aqueous hydrochloric acid and mor- pholine in acetone could be used in the procedure. Phthalic anhydride was purified by recrystallization from anhydrous ethyl ether. This sample, analyzed by the morpholine method, had a purity of 102%. -71- A fresh supply of TMA was then obtained through the courtesy of A. F. Schubert of Amoco Chemicals Corporation. of 99.1%. Analysis by. the morpholine method indicated a purity It thus appeared that the morpholine method would be acceptable for determining anhydride content in the presence of carboxyl groups. The anhydride reacts mole for mole with morpholine to give an amide and a carboxyl group. The carboxyl groups may form salts with the morpholine, but these would be titratable. The method was also tested on diethyl phthalate, a mixture of monomethyl trimellitate and trimellitic acid, and a sample of trimethyl trimellitate which had been prepared with a sulfuric acid catalyst by a procedure similar to that given in an Amoco Technical Data Sheet (3). Since none of these materials reacted with morpholine, the method was shown to be suitable for the determination of anhydrides without interference from acids or esters. -72- APPENDIX II DETERMINATION OF MOLECULAR WEIGHT The molecular weights of the trifluoroacetyl-TMA unsymmetrical anhydride, the acetyl-TMA unsymmetrical anhydride, the acid chloride of TMA, bis(trimellitic anhydride) anhydride, and 4-carbomethoxy phthalic anhydride were determined by freezing point depression as outlined bySteinbach and King (89). determinations were made with benzene as the solvent. All but one of the Bis(trimellitic anhydride) anhydride is insoluble in benzene; its molecular weight was determined from the freezing point lowering of dioxane. A Beckmann thermometer graduated into 0.01°C. divisions was employed. The solvent or solution was transferred to a test tube which was placed within a larger test tube to separate it from the cooling bath (ice water). and a ring stirrer were placed in the solution. The thermometer The freezing points were then determined by recording the temperatures at thirty-second intervals to obtain the cooling curves. By obtaining the freezing point lowering between solvent and solu- tion (AT) and knowing the molal freezing point lowering constant for the solvent (Kf), the molecular weight was calculated by: Mol. wt. = (l000)(wt. of solute)(Kf) / (wt. of solvent)(AT). -73APPENDIX III FLUORINE ANALYSIS The method employed for fluorine analysis consisted of a Schoniger combustion (90) followed by titration with cerous chloride (91-92). The modified method con- sisted of weighing a sample (10 to 30 mg.) onto a piece of filter paper with about 20 mg. sodium peroxide. The filter paper was then rolled and placed in the plati- num holder of the stopper of the Schoniger flask. Distilled water (10 ml.) was added to the flask to absorb the combustion products. displaced with oxygen. The air in the flask was The paper was ignited and quickly placed in the flask. An interval of 30 minutes was allowed for complete absorption of combustion products. After opening the flask, the solution was acidified with 5 ml. 0.01N hydrochloric acid and the carbon dioxide was boiled off. The solution was neutral- ized with O.O1N sodium hydroxide using bromthymol blue as an indicator and then made slightly acidic (pH 5 to 6). Ethanol (10 ml.) was added and the fluoride was then titrated with 0.01N cerous chloride using a Murexide indicator. A blank was also run, The original method did not include the addition of the catalyst, which was acceptable when a known compound was tested. Perfluorocaprylic acid was recrys- tallized from carbon tetrachloride and then from toluene. obtained were: 69.4, 70.9% F (theoretical: The analytical results 68.83%). When a freshly prepared sample of the unsymmetrical anhydride of TMA and TFAA was analyzed by the Schoniger technique, low results were obtained: 11.6% F (theoretical: 19.78% F). 10.1, It appeared probable that some disproportiona- tion was taking place before combustion was completed, and that some oxidation aid might be necessary. Opinion in the literature is divided on the need for catalysts -74in the combustion of fluorinated compounds. Rogers and Yasuda (93) employed the Schoniger combustion and reported no difficulty in compounds containing the -CF3 group. Johnson and Leonard (94) also stated that decomposition of the -CF3 group by flask combustion is complete and that no oxidation aids are necessary. However, Belcher, et al. (95) observed that some compounds containing the -CF3 group gave low results, and attributed this to the formation of carbon tetrafluoride. Belcher obtained improved results with the aid of potassium chlorate. Steyermark, et al. (96) and Senkowski, et al. (97), also observed the need for a catalyst and employed sodium peroxide. The analysis was repeated with the addition of about 20 mg. sodium peroxide. With the aid of the catalyst, the results were quite satisfactory: (theoretical: 19.78% F). 19.3, 19.5% F -75APPENDIX IV ESTERIFICATION REACTIONS OF ALCOHOLS WITH THE TRIMELLITIC ANHYDRIDE-TRIFLUOROACETIC ANHYDRIDE SYSTEM ESTERIFICATION OF THE UNSYMMETRICAL ANHYDRIDE OF TMA AND TFAA Several reactions were conducted by adding an alcohol to the unsymmetrical anhydride. In this state, the solid anhydride reacts to yield primarily TMA and the trifluoroacetate ester. Phenol After two hours at room temperature, a mixture of TMA (5.0 g., 0.026 mole) and TFAA (6.75 g., 0.032 mole) solidified. After the addition of 2.289 g. phenol (0.024 mole) the mixture was heated at 75° and gradually liquefied. hours the mixture had partially solidified. After twenty Filtration gave a yellow-white solid which was washed with two 25-ml. portions of carbon tetrachloride. The white crys- talline solid remaining in a yield of 3.735 g. was TMA, and had a m.p. of 162-165 ° and anhydride equivalent of 195 (theoretical: 168° and 192.1). Additional TMA (0.48 g.) separated on addition of the carbon tetrachloride washings to the original filtrate, m.p. 152-161 ° and anhydride equivalent: 191. Butanol-1 A mixture of TMA (10 g., 0.0522 mole) and TFAA (12 g., 0.0572 mole) was allowed to react at room temperature. Initially, the TMA appeared to be dissolving, but after standing overnight, the mixture had completely solidified. On heating on a water bath at 75° , the mixture partially melted and then solidified. would not liquefy on continued heating at this temperature. The mixture Butanol-1 (3.888 g., 0.0524 mole) was added to this mixture, which was then heated at 75° for 8.5 hours. -76The mixture was then extracted with 30 ml. boiling anhydrous ethyl ether and filtered. The ether-insoluble material, recovered in 8.224 g. yield, was probably mostly TMA, m.p. 156-182° and anhydride equivalent 203 (expected values: 192.1). 168 ° and Several small additional crops which were obtained on refrigeration and concentration were not resolved. The ether was stripped from the extract and the remaining liquid was distilled to yield 6.15 g. of product which gave a positive ferric hydroxamate test and negative test for alcohols with the ceric nitrate reagent. This liquid should be n-butyl trifluoroacetate and had a saponification equivalent of 210 and n0 1.3288 (theoretical: 170 and nD 1.3394)(72). Methanol Several reactions were carried out between methanol and the unsymmetrical anhydride of TMA and TFAA. The first reaction was an attempt to obtain 4-carbo- methoxy phthalic anhydride. The unsymmetrical anhydride was prepared by refluxing a solution of TMA (5 g., 0.026 mole) in TFAA (30 g., 0.143 mole) for five hours. The cooled solu- tion was then filtered and the filtrate solidified on cooling. This product was dried in vacuo at room temperature to give 6.457 g. (0.0224 mole) 4-trifluoroacetyl TMA, m.p. 93-96, 220-221 ° (86.1% yield). Methanol (0.72 g., 0.0225 mole) was added dropwise to the unsymmetrical anhydride with evolution of heat. The product was extracted with 10 ml. cold anhydrous ether, filtered, and washed with a second portion of ether. 152-163 ° . Sublimation at 85° and 1-2 mm. led to a trace of material having a m.p. of 146-168 ° . 145-163° . The ether-insoluble material (1.97 g.) had a m.p. of The ether washings were concentrated to dryness, 0.34 g., m.p. Sublimation again led to a small amount of material, m.p. 139-155 °. I -77° The ether extract was also concentrated to dryness to yield 1.43 g., m.p. 115-163 . Sublimation for three days at 85 ° and 1-2 mm. removed only a trace of material. ° This product softened slightly at 88° but did not melt until 150 . It is possible that this fraction contained a trace of 4-carbomethoxy phthalic anhydride, but the major portion of the sublimate was probably TMA. Since the ester anhydride was not obtained, it was necessary to prove the formation of methyl trifluoroacetate. The unsymmetrical anhydride was prepared as above, on a larger scale, and recrystallized from benzene to give 21.1 g. (0.073 mole), m.p. 93-95, 221 ° . wise with evolution of heat. 43-45 ° . Methanol (3.96 g., 0.124 mole) was added drop- A colorless liquid (2.6 g.) was distilled with b.p. 28 This liquid gave a positive ferric hydroxamate test and had nD 20 Literature values for methyl trifluoroacetate are b.p. 43° and n0 1.3346. 1.29073 (75). An infrared spectrum compared favorably with the known spectrum of methyl trifluoroacetate but had several extra bands. Absorption at 678 and 1485 cm. indicated a benzene impurity while absorption at 1032 and 3360 cm. a methanol impurity. -1 indicated It was decided to repeat this experiment without adding quite as much excess methanol and taking greater care to remove all benzene after recrystallizing the unsymmetrical anhydride. In this case, methanol (3.01 g., 0.094 mole) was added dropwise to the re- The fraction of crystallized unsymmetrical anhydride (25.8 g., 0.0896 mole). distillate of this product having b.p. 43-44 ° was retained. based on methyl trifluoroacetate). This product had n for methyl trifluoroacetate are b.p. 43° and n 0.5 g. having b.p. 44-46 ° was also 3 Yield: 233 retained, n 1.293. Literature values 1.293. 1.29073 (75). 1 3.0 g. (26.2% A second cut of Infrared spectra of these two fractions matched the known spectrum of methyl trifluoroacetate. Fig. 2 for the spectrum of the isolated methyl trifluoroacetate). (See I! -79An additional reaction was carried out to determine the effect of excess methanol. TMA (1.0 g., 0.0052 mole) was heated in 27 g. TFAA (0.128 mole) in an enclosed glass tube. lation. Some excess TFAA (22 g., 0.105 mole) was removed by distil- Frothing and heat evolution accompanied the addition of 1.58 g. methanol (0.0493 mole). Two precipitates were recovered from the reactant solution. The first product had a m.p. of 148-151 ° with a neutralization equivalent of 71.25 and an anhydride equivalent of 285. ester. This was probably a mixture of TMA and an The second product was a mixture of 1- and 2-methyl trimellitate, with a neutralization equivalent of 109.5 (theoretical: 112.1) and no anhydride content. The mixture softened at 179 ° , m.p. 200-206 ° . ACYLATION REACTIONS OF TMA AND TFAA IN ACETONE Different solvents were examined as media for the esterification of the TMATFAA system. Acetone was used in several reactions; the main reactions are described below. Butanol-1 TMA (10 g., 0.052 mole) and TFAA (13.5 g., 0.064 mole) were dissolved in 40 ml. acetone. After two hours at room temperature, 3.858 g. n-butyl alcohol (0.052 mole) were added to the yellow solution, with noticeable evolution of heat. After 72 hours, a small amount of a brown flaky precipitate settled and was filtered off. After a week, no further precipitation occurred. and four fractions were collected. The solution was distilled The low boiling fraction was acetone, while the last three fractions all gave positive ferric hydroxamate tests and contained n-butyl trifluoroacetate (refractive index at 18°: alent: 170) (72). 1.3394, saponification equiv- The indices of refraction at 27° for these fractions before and after charcoal treatment were, respectively: 1.3490, 1.3502; 1.3339, 1.3348; -80- and 1.3250, 1.3264. The saponification equivalents of the last two fractions were 142 and 163. Phenol TMA (25 g., 0.130 mole) was dissolved in 60 ml. acetone and 33.75 g. TFAA (0.160 mole). After two hours, 11.57 g. phenol (0.123 mole) were added. standing at room temperature, three successive crops crystallized. On These were filtered, washed with acetone, and dried. After standing five months, the fourth crop was filtered off, washed, and dried. The first two crops were recrystallized from 2:1 acetone:methyl ethyl ketone. A brief summary of the properties of these products is presented in Table V. TABLE V 'ANALYSIS OF TMA-TFAA-PHENOL REACTION PRODUCTS ° Product Yield, g. 1 1.0 222-226 124 2 1.4 217-221 127 3 2.2 148-156 4 13.8 m.p., C. Anhyd. Equiv. Neut. Equiv. 60.4 148-155,.210 The first two crops were bis(trimellitic-anhydride) anhydride while the latter two crops contained a large amount of TMA. The fourth product (brown crystals) had a phenolic odor and was extracted with hot 1,2-dichloroethane. The insoluble fraction was trimellitic acid, m.p. 222°d. obtained from the extract. Four crops were The first crop was TMA, yield 7.7 g. and m.p. 165-169 ° . The second crop (0.96 g.) had m.p. 147-153 ° . This was mainly TMA; after three re- crystallizations, yield: 0.7 g. and m.p. 167-169 ° . identified in the third crop (0.5 g.). TMA was the only-pure product The final crop, resulting from concentra- tion of the remaining extract to dryness, was probably impure phenol, m.p. 35-38 ° . -81- Cresol The reaction of TMA, TFAA, and o-cresol in acetone followed the pattern of the above reaction with phenol. TMA (25 g., 0.130 mole) was dissolved in 60 ml. acetone and 33.75 g. TFAA (0.160 mole). cresol (0.124 mole) were added. obtained. After two hours, 13.4 g. redistilled o- As in the previous reaction, four crops were The first two crops were recrystallized from acetone. Their properties are indicated in Table VI. TABLE VI ANALYSIS OF TMA-TFAA-CRESOL REACTION PRODUCTS Product Yield, g. m.p., °C. Anhyd. Equiv. 1 0.84 221-225 124 2 3.05 221-225 124 3 1.25 162-215 13.7 4 140-150 Again, the first two crops were bis(trimellitic anhydride) anhydride (anhydride equivalent: 122.08) while the latter two crops were mixtures. After recrystallizing the fourth crop three times from o-dichlorobenzene, it had a m.p. of 164-167° and anhydride equivalent 227. TMA with a small impurity. is 192.1. This product (10.41 g.) was probably The m.p. of TMA is 168 ° while the anhydride equivalent (The anhydride equivalent of the ester anhydride of TMA and o-cresol would be 282.24.) A fourth recrystallization brought the m.p. to 164-166 ° and the anhydride equivalent to 210. Absence of Alcohol If the initial product obtained from reactions in acetone is bis(trimellitic anhydride) anhydride, then this disproportionation product should also result if -82- no alcohol is present. Although later work indicated this trianhydride could be obtained most easily by refluxing the two anhydrides in acetone, the early work was performed at room temperature. TMA (50 g., 0.26 mole) dissolved in 160 ml. acetone and 67.5 g. (0.321 mole) TFAA. Over the course of several days, four similar crops were filtered from this mixture. The first three crops were recrystallized from acetone while the fourth crop was recrystallized from o-dichlorobenzene. ials are presented in Table VII. These products should all be bis(trimellitic anhydride) anhydride (anhydride equivalent: 61.04). Analyses of the purified mater- 122.08 and neutralization equivalent: The stability was estimated by storing a sample of the third crop in a desiccator over phosphorus pentoxide. unchanged. After eighteen days the melting point was An infrared spectrum obtained on a potassium bromide disk (Fig. 3) indicated carbonyl absorption at 1860 and 1776 cm. . These bands are present in TMA and have been attributed to the cyclic anhydride ring. absorption at 1801 and 1737 cm. anhydride group. There was also carbonyl ; these bands are attributed to the open chain There was also an indication of hydroxyl group activity, which is inconsistent with the proposed structure. ture adsorbed by the potassium bromide. However, this could be due to mois- Two spectra run in hexachlorobutadiene and one in nujol indicated that no hydroxyl groups, and therefore no carboxyl groups, were present. The infrared spectra of TMA and trimellitic acid, Fig. 3, are presented for comparison. TABLE VII ANALYSIS OF TMA-TFAA-ACETONE REACTION PRODUCTS ° Product Yield, g. 1 3.6 217-220 124 2 4.4 223-227 125 3 3.1 222-225 126 4 14.0 220-224 .124 m.p., C. Anhyd. Equiv. Neut. Equiv. 60.9 l1) -84The reaction was repeated on a slightly smaller scale to obtain a fresh sample of bis(trimellitic anhydride) anhydride for elemental analysis. crystallized from 1,2-dichloroethane and had a m.p. of 224-227° . gen analyses were obtained by the Geller Laboratories. The sample was reCarbon and hydro- Their results along with oxygen content, by difference; anhydride equivalent; neutralization equivalent; and molecular weight are presented with the theoretical values in Table VIII. VIII ANALYSIS OF BIS(TRIMELLITIC ANHYDRIDE) ANHYDRIDE C, % H, % 0, % Found 58.60 2.02 39.38 128 59.5 356+10 Calc. 59.03 1.65 39.32 122 61.0 366.23 Anhyd. Equiv. Neut. Equiv. Mol. Wt. ESTERIFICATION IN TRIFLUOROACETIC ACID In previous studies of esterifications of carboxylic acids promoted by TFAA, increased yields of the carboxylic acid esters were obtained when the reactions were carried out in trifluoroacetic acid. For this reason a series of reactions with various alcohols were conducted in this medium. Methanol One reaction with methanol was carried out in trifluoroacetic acid followed by sublimation, which should be the most efficient method of removing any'4-carbomethoxy phthalic anhydride. TMA (5.0 g.), TFAA (5.467 g.), and trifluoroacetic acid (29.68 g.), molar ratio 1:1:10, were refluxed to effect solution and formation of the unsymmetrical anhydride. The solution was then cooled and 0.834 g. methanol (equimolar to TMA) was added with heat evolution. at room temperature for 40 hours. The mixture was allowed to react The precipitate which had formed (0.25 g.) had -85a m.p. of 157-190°. Sublimation at 800 and 1-2 mm. led to a small amount of impure TMA, m.p. 146-158°. The remaining solution was concentrated to dryness in vacuo at room temperature. The dried product (3.51 g.) had a m.p. of 151-163. tion removed 0.02 g. , m.p. 110-160°. Sublima- No ester was isolated. Butanol Several reactions were carried out with butanol-2 in trifluoroacetic acid. However, the ester anhydride of TMA was never positively identified. conditions were varied as shown in Table IX. Reactant TMA was the only product positively identified from each reaction. TABLE IX ESTERIFICATION IN TRIFLUOROACETIC ACID Reaction TMAa 23 0.0522 0.0522 0.468 0.0521 25 120 28 0.0522 0.0522 0.104 0.0513 25 72 30 0.0522 0.0523 0.351 0.0522 85 8 31 0.0522 0.0523 0.351 0.0522 85 24 TFAA a TFAcidab Butanola Temp., °C. Time, hr. aMolar quantities. Trifluoroacetic acid. Crystalline products were obtained on standing, while vacuum concentration led to the isolation of secondary crops. Recrystallization was attempted from various solvents (acetone, methyl ethyl ketone, dioxane, o-dichlorobenzene, ether, and 1,2-dichloroethane) but TMA or a mixture was the usual result. One unusual product was obtained as the third crop from Reaction 23. After three recrystallizations from o-dichlorobenzene, the crystals recovered had a -86m.p. of 166-167° and an anhydride equivalent of 250. This represents a m.p. close to TMA (168° ) and an anhydride equivalent close to the butyl ester anhydride (248.226). Duplicate equivalent determinations were carried out by dissolving the samples in hot water and titrating with sodium hydroxide. obtained. 82.7. A value of 79.8 was The saponification equivalent of the butyl ester anhydride would be An equivalent of 92.7 was obtained when the sample was titrated in aqueous ethanol. An infrared spectrum exhibited carbonyl absorption at 1860 and 1773 cm. (also present in TMA due to the cyclic anhydride ring). differences from TMA, however. The spectrum had several Carbonyl absorption was also shown at 1705 cm. Small-scale tests for the presence of the sec-butyl group were inconclusive. . One saponified sample was distilled and the first few milliliters of distillate gave a negative test for alcohols with the eerie nitrate reagent. theoretical concentration also gave a negative test. was extracted with benzene. A blank of the same A second saponified sample The benzene extract was reacted with acetyl chloride. After hydrolysis of the excess acetyl chloride, the sample was tested with ferric hydroxamate for sec-butyl acetate. A very weak test was given. Duplicate carbon and hydrogen analyses were run on a sample by the Geller Laboratories. The results along with the oxygen content (by difference) are presented in Table X along with the theoretical values for TMA and the butyl ester anhydride. In spite of the sharp melting point, the sample must have been a mixture. TABLE X ELEMENTAL ANALYSIS OF PRODUCT 23 Sample C, % H, % 23 (1) 58.96 2.91 (2) 59.32 2.71 Av. 59.14 2.81 38.05 4-Butyl TMA 62.90 4.87 32.23 TMA 56.26 2.10 41.64 O, % -87In addition to the usual recrystallization techniques, one attempt was made to purify the main product from Reaction 31 by extraction. After recrystallizing this product from o-dichlorobenzene the sample had an anhydride equivalent of 212. This value is slightly higher than TMA. If this sample contained a small amount of the butyl ester anhydride, it seemed possible to extract the TMA with sodium bicarbonate. This procedure might not extract the ester anhydride. 4.4 g. were dissolved in 300 ml. 1,2-dichloroethane. Consequently, The solution was filtered and extracted with three 50-ml. portions of 5% sodium bicarbonate. The organic solution was washed with distilled water, dried with anhydrous magnesium sulfate, filtered, and concentrated to 7 ml. When there was no crystallization, the solu- tion was concentrated almost to dryness, but only a trace of material crystallized, m.p. 167-168.5 ° (TMA). The sodium bicarbonate solution was acidified with hydro- chloric acid with no precipitation. This solution was then extracted with three 40-ml. portions of 1,2-dichloroethane. After washing, drying, and filtering, the organic extract was concentrated almost to dryness with no crystallization. If there were any anhydride ester in this sample, it must have been completely extracted by the sodium bicarbonate solution. An additional reaction of TMA, TFAA, and butanol-2 was carried out in trifluoroacetic acid to determine the effect of the addition of excess alcohol. The TMA (10 g., 0.0522 mole) dissolved in 40 g. (0.351 mole) trifluoroacetic acid and 11 g. (0.0523 mole) TFAA after three hours. g., 0.104 mole) was then added. allowed to cool slowly overnight. The sec-butyl alcohol (7.72 The mixture was refluxed for six hours and then The white powdery precipitate (8.64 g.) was filtered, washed with benzene, and dried, m.p. 224-240°d. This product had a very low anhydride content, and was extracted with 150 ml. hot 1,2-dichloroethane to remove the anhydride. Only 0.05 g. was recovered from the solution, m.p. 167-190 °. -88The insoluble white powder (7.94 g.) was recovered, m.p. 235-242°d. This product gave a weak ferric hydroxamate test and had an equivalent weight of 71.4. sample was dissolved in p-dioxane for recrystallization. gave a weak ferric hydroxamate test, m.p. 242-244°d. The The first crop (4.2 g.) The equivalent weight was determined by dissolving three samples in hot water and titrating with sodium hydroxide: 88.7, 89.2, 89.1 (89.0). This value is the same as the saponification equivalent for a mono-butyl trimellitate (88.75). The product was probably the 1-butyl or 2-butyl derivative (or a mixture of the monoesters). An infrared spectrum exhibited a strong similarity to trimellitic acid but also gave an indication of an ester group at 1733 cm. 1705 cm. in addition to carboxyl absorption at -1 Other Alcohols Two additional reactions were conducted in trifluoroacetic acid, one with a longer chain secondary alcohol (capryl) and another with phenol. In the first reaction, TMA (10 g., 0.0522 mole) was dissolved in TFAA (10.94 g., 0.0522 mole) and trifluoroacetic acid (53.4 g., 0.458 mole). The solution was filtered through a fine sintered glass filter and 6.77 g. capryl alcohol (0.052 mole) were added. The solution turned orange, accompanied by the evolution of heat, and crystallization started immediately. After standing overnight, 7.24 g. TMA were filtered off, m.p. 166-168 ° and anhydride equivalent 210 (theoretical 168 ° and 192.1). Small secondary crops which separated on refrigeration gave negative ferric hydroxamate tests after recrystallization from dioxane. In the second reaction, 11 g. (0.0523 mole) TFAA and 40 g. (0.351 mole) trifluoroacetic acid were added to 10 g. (0.0522 mole)-TMA. After three hours with gentle warming the TMA dissolved and 4.63 g. (0.0492 mole) phenol were added. ( -89- The solution was refluxed three hours (830) and then allowed to cool slowly overnight. The 3.21 g. of white crystals which separated had a m.p. of 164-168° and an anhydride equivalent of 203. After three recrystallizations from o-dichloro- benzene, the sample had a m.p. of 165-167° and anhydride equivalent: On refrigeration, 4.63 g. white crystals separated. 207 (TMA). This second crop had an anhydride equivalent of 216, softened at 70-80 ° and melted at 151-158 ° . sample was recrystallized from o-dichlorobenzene, m.p. 158-168° . acetic acid odor was noticed on filtering. The A trifluoro- After three subsequent recrystalliza- tions, the m.p. was 163-166° and anhydride equivalent: 209 (TMA). The third crop of white crystals (1.61 g.) which separated was similar to the second crop, m.p. 80-162 ° and anhydride equivalent: 216. Again, on recrystallization from o-di- chlorobenzene, a trifluoroacetic acid odor was noticeable. After three recrystal- lizations, the m.p. was 165-166° and anhydride equivalent: 209. After concentra- tion of the o-dichlorobenzene filtrate, 0.022 g. crystals separated which softened at 139 ° and melted at 159-161 ° . The remaining reactant solution was concentrated almost to dryness. material (3.26 g.) was filtered and washed with benzene. odor and melted over 105-170 ° . 1,2-dichloroethane. The solid The product had a phenolic This fourth product was extracted with 50 ml. hot The small amount of insolubles decomposed over 170-225 ° and gave a weak ferric hydroxamate test. crystallized into three crops. The dichloroethane solution was fractionally The first crop, after recrystallization, had a m.p. of 167-168.5 ° and an anhydride equivalent of 205 (TMA). The second crop, after three recrystallizations, had a m.p. of 164-168 ° and an anhydride equivalent of 202 (TMA). The third crop finally crystallized from a viscous solution. This crop, m.p. 98-120 ° , had a phenolic odor, and was recrystallized from dichloroethane. The first crop (0.02 g.) had a m.p. of 158-159.5 °. An infrared spectrum -90- indicated carbonyl absorption at 1705 cm. 1748 cm. , probably due to ester groups. -1 , probably due to carboxyl groups, and This subcrop is probably a monophenyl trimellitate. ESTERIFICATION IN ADDITIONAL SOLVENTS A number of esterification reactions of the TMA-TFAA system were carried out in solvents other than acetone and trifluoroacetic acid. Solvents which were briefly investigated were chloroform, ether, carbon tetrachloride, o-dichlorobenzene, and 1,2-dichloroethane. Chloroform One reaction between the unsymmetrical anhydride and butanol-l was conducted in chloroform. Butanol-l (0.405 g., 0.0055 mole) was added to the unsymmetrical anhydride (1.129 g., 0.0039 mole) followed by 4 ml. chloroform. refluxed thirty minutes. The mixture was After cooling slowly, the mixture was poured into an excess of 5% sodium bicarbonate. The organic layer was separated, washed with 5% sodium bicarbonate, and washed with distilled water. The alkaline solution was extracted with chloroform and the chloroform solutions were combined, washed with water, and dried with anhydrous magnesium sulfate. Evaporating to dryness showed that the chloroform had not extracted a significant amount of material. The aqueous solution was acidified with dilute hydrochloric acid, resulting in the precipitation of a product with m.p. 164-172° , neutralization equivalent: 133, and no anhydride content. Refrigeration of the filtrate precipitated an additional product of m.p. 174-176° and neutralization equivalent: 129. These products are probably mono- butyl trimellitates (neutralization equivalent: 133.12). to trimellitic acid, neutralization equivalent: Further concentration led 70.98 (theoretical: 70.05). This sample decomposed at 234-237 ° , indicating the formation of the cyclic anhydride -91ring and evolution of water, and remelted at 166-167 ° on subsequent reheating (TMA has m.p. 168°). Ether A similar reaction was carried out in anhydrous ethyl ether. A reactant mixture of 2.63 g. (0.0091 mole) of the unsymmetrical anhydride and 0.676 g. (0.0091 mole) butanol-l was diluted with 10 ml. ether. Reaction at room tempera- ture led to the crystallization of 0.82 g. TMA (m.p. 165-168°). The remaining solution was treated in a manner analogous to the treatment of the chloroform solution of the previous reaction. The solid products obtained were impure. The ether was stripped from the solution leaving a liquid with a slight yellow 22 coloration, n 2 1.3560. 18 nD This liquid was probably impure n-butyl trifluoroacetate, 1.3394 (72). Other Chlorinated Solvents Three reactions were carried out in other solvents: o-dichlorobenzene, and 1,2-dichloroethane. carbon tetrachloride In the first reaction, 6.75 g. (0.032 mole) TFAA were added to 5.0 g. (0.026 mole) TMA. The mixture solidified after several hours at room temperature, and would not liquefy on heating at 75 ° . The mixture was heated at 75 ° for 32 hours after the addition of 20 ml. carbon tetrachloride and 2.323 g. (0.025 mole) phenol. The first two crops to separate (4.0 g.) were both TMA, m.p. 165-167 ° and anhydride equivalent: 200 (theoretical 168 ° and 192.1). This accounted for 80% of the starting material. A small additional crop (0.2 g.) separated on cooling and was recrystallized from anisole. This product had an anhydride equivalent of 256, which is close to that of the phenyl ester anhydride (268.2). However, the m.p. indicated the sample was impure. sample softened at 114-117 ° and melted at 156-158°.) (The -92- Two reactions were carried out between the TMA-TFAA system and butanol-2 in The reactions are summarized in Table XI. 100 ml. solvent. TABLE XI REACTIONS IN CHLORINATED SOLVENTS Reaction TMAa 33 0.0521 34 0.0521 TFAAa Butanola Solvent 0.0785 0.0521 o-dichlorobenzene 0.0785 0.0521 1,2-dichloroethane aMolar quantities. Most of the TMA was recovered from these reactions. TMA was the only product positively identified (by melting point and anhydride equivalent). -93APPENDIX V PREPARATION OF 4-CARBOMETHOXY PHTHALIC ANHYDRIDE FROM TMA AND DIAZOMETHANE A sample of 4-carbomethoxy phthalic anhydride was desired for identification purposes of the esters obtained from reactions of methanol with the anhydride derivatives of TMA. Duff, et al. (2) reported the preparation of this ester from the reaction of TMA and diazomethane but gave little experimental detail. The ethereal diazomethane solution was prepared as outlined by Weygand (98). Nitrosomethylurethane (12 ml.) in 480 ml. ether was reacted with 14.4 ml. 25% aqueous potassium hydroxide. The yellow distillate was ethereal diazomethane. Nine reactions of varying time and temperature were carried out between TMA and ethereal diazomethane. Only low yields of the anhydride ester could be obtained. The products appeared to be mainly mixtures. diazomethane was added to TMA (4.5 g.). In one reaction, an excess of ethereal Reaction was indicated by gas evolution and a decrease in intensity of the yellow coloration of the solution. After con- centration to dryness the product was extracted with benzene at room temperature. Most of the product (4.48 g.) was insoluble, m.p. 141-210°d. benzene extract led to a yellow sirup. Concentration of the Sublimation of this sirup at 80° and 1 to 2 mm. led to the isolation of about 0.2 g. crude 4-carbomethoxy phthalic anhydride (4.1% yield), m.p. 86-101°. After low temperature recrystallization from ether, the m.p. was 99-102°. Although anhydrous ethyl ether was employed, it would seem that part of the product might have been hydrolyzed. It also appears that the cyclic anhydride ring is not as stable to diazomethane as might be implied by the reported preparation (2). Bhati (99) recently reacted succinic anhydride with diazomethane and obtained a diazo-keto-ester to which he assigned the structure: -94- CH2 -CO-CH-N=N CH2 -COOCH3 In addition, with substituted phthalic anhydrides, Bhati obtained keto-lactones of the type: Aiat0 R A reaction of this type in the case of TMA and diazomethane might account for the low yield of 4-carbomethoxy phthalic anhydride. -95APPENDIX VI METHYLATION OF THE ACID CHLORIDE OF TRIMELLITIC ANHYDRIDE Several additional reactions were conducted between the acid chloride of TMA and methanol. The rate of addition of methanol to the benzene solution of the acid chloride was varied in two reactions, while vacuum was applied in a third reaction to facilitate the removal of the hydrochloric acid. Other reactions were carried out between the solid acid chloride and methanol, and with methanol and the acid chloride in various solvents (1,2-dichloroethane, ethyl ether, and toluene). REACTION 5 The acid chloride (4.0 g., 0.019 mole) was dissolved in 1,2-dichloroethane at room temperature and methanol (0.594 g., 0.019 mole) was added. chloride was evolved on gentle warming of the solution. separated, m.p. 202-214 ° . Hydrogen On refrigeration, 0.1 g. The solution was eventually concentrated to a sirup. Cooling to -80° followed by slow warming and recooling did not bring about crystallization. After about seven weeks, a white precipitate was observed in the sirup, which was diluted with 10 ml. dichloroethane and filtered. The dried product (0.24 g.) had a m.p. of 195-213 ° . The sample gave a positive ferric hydroxamate test and was extracted with hot benzene. The insoluble material had a m.p. of 207-215 ° and gave only a weak ferric hydroxamate test. Nothing crystallized from the benzene solution, which on concentration led to a sirup. The dichloroethane filtrate was concentrated again to a sirup. Sublimation at 60° and 2 mm. led to the collection of 0.02 g. of 4-carbomethoxy phthalic anhy- dride, m.p. 87-94°. was 99-101 ° . After low-temperature recrystallization from ether, the m.p. -96REACTION 6 This reaction was similar to the first reaction: 7.87 g. acid chloride (0.0374 mole) and 1.188 g. methanol (0.0371 mole) in benzene. This time, however, the solution was placed under vacuum immediately to facilitate removal of hydrogen chloride. Concentration and refrigeration failed to initiate crystallization and the sample was finally concentrated to a sirup. Alternate cooling to -80° followed by slow warming with small additions of ether and scratching failed to bring about crystallization. REACTION 7 This reaction of 5.22 g. acid chloride (0.0248 mole) and 0.792 g. methanol (0.0247 mole) was also carried out in benzene. In this case, the methanol was added dropwise with agitation at a rate of 0.01 ml./min. temporarily over potassium hydroxide. The solution was stored Refrigeration, cooling, and scratching failed to initiate crystallization and the solution was eventually concentrated to a sirup. After six weeks, a precipitate was observed in the sirup, which was then diluted with benzene and filtered. The dry product (0.87 g.) had a m.p. of 120- 145° and was extracted with hot benzene. benzene extract, m.p. 125-158° . centration. The remaining benzene extract was a sirup on con- The fraction insoluble in benzene (0.53 g.), m.p. 147-170° , was extracted with 1,2-dichloroethane. 215°d. On cooling, 0.1 g. separated from the Trimellitic acid (0.1 g.) was insoluble, m.p. Three crops were obtained from the dichloroethane extract. The first crop (0.06 g.) had m.p. 213-217°, while the second (0.15 g.) had a m.p. of 166-168° (TMA). The third crop (0.04 g.) had a m.p. of 132-146 ° and a m.p. of 142-152° after recrystallization from dichloroethane. -97A second product (0.2 g.) separated from the benzene filtrate, m.p.. 109-190°d. Two crops obtained from a dichloroethane extract of this product had a m.p. of 184-213°d. (0.03 g.) and 179-198°d. (0.1 g.). An additional product separated from the benzene solution of the sirup from this reaction, m.p. 95-180 ° . Sublimation of this product at 70 to 80° and 1 to 2 mm. led to the isolation of 0.109 g. crude 4-carbomethoxy phthalic anhydride. On recrystallization from ether the pure anhydride ester was obtained, m.p. 100-102° . REACTION 8 The acid chloride (4.93 g., temperature. began. 0.0234 mole) was dissolved in ether at room The solution was cooled to -40° , at which point crystallization The mixture was warmed to 0° to effect solution, and 0.713 g. methanol (0.0223 mole) were added. perature for five hours. The solution was cooled to -40° and kept at this temThe crystallized product (2.13 g.) was filtered off and shown to be the acid chloride, m.p. 67°. The remaining ether solution was concen- trated, leading to a yellow sirup. REACTION 9 An additional reaction was conducted in benzene. The acid chloride (2.13 g., 0.01 mole) was dissolved in 20 ml. benzene at room temperature. A solution of 0.317 g. methanol (0.01 mole) in 10 ml. benzene was placed in a separatory funnel and added dropwise to the refluxing solution over a period of four hours. The solution was refluxed an additional two hours, cooled, concentrated to 15 ml., and set aside. No precipitation occurred after two weeks and the solution was even- tually concentrated to a sirup. The sirup was then dissolved in anhydrous ether. On standing, 0.45 g. separated, m.p. 120-170 ° . The ether filtrate was cooled to -98-80° and 0.22 g. separated, m.p. 108-156° . Sublimation of this product at 80° and 1 to 2 mm. led to the crude 4-carbomethoxy phthalic anhydride, m.p. 88-101°, which was purified by recrystallization from ether, m.p. 99-101° . REACTION 10 The acid chloride (2.4 g., 0.011 mole) was dissolved in 50 ml. toluene and cooled to -35° , at which point 0.43 g. (0.013 mole) methanol were added. The solution was cooled to -50° and maintained at that temperature for seven hours, after which it was filtered and concentrated to 25 ml. Cooling to -75° then led to the crystallization of 1.27 g. unchanged acid chloride, m.p. 63-66 ° . Further cooling at -75° for 24 hours led to additional crystallization of the acid chloride, m.p. 64-66 ° . Further concentration of the toluene filtrate led to a small amount of a dark sirup. The recovered acid chloride (1.27 g., 0.006 mole) was redissolved in 2 ml. toluene at room temperature and 0.158 g. (0.005 mole) methanol were added. Concentration of this solution also led to a sirup. REACTION 11 Methanol (0.158 g., 0.005 mole) was added dropwise to the solid acid chloride (1.6759 g., 0.008 mole). Hydrogen chloride was evolved. The product was extracted with 10 ml. cold ether and 0.1 g. unchanged acid chloride was filtered off, m.p. 61-64° . The ether extract was cooled to -75° leading to the crystallization of 0.58 g., m.p. 53-61 ° . This was extracted with cold benzene. product (0.02 g.) had a m.p. of 91-105°. phthalic anhydride. The recrystallized This should be the impure 4-carbomethoxy The remaining benzene extract was a sirup on concentration.