Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Vectors in gene therapy wikipedia , lookup
Designer baby wikipedia , lookup
Maximum parsimony (phylogenetics) wikipedia , lookup
Point mutation wikipedia , lookup
History of genetic engineering wikipedia , lookup
Quantitative comparative linguistics wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Transposable element wikipedia , lookup
Minimal genome wikipedia , lookup
Whole genome sequencing wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Gene expression profiling wikipedia , lookup
Koinophilia wikipedia , lookup
DNA barcoding wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Genomic library wikipedia , lookup
Non-coding DNA wikipedia , lookup
Human Genome Project wikipedia , lookup
Human genome wikipedia , lookup
Pathogenomics wikipedia , lookup
Genome evolution wikipedia , lookup
Microevolution wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Helitron (biology) wikipedia , lookup
Genome editing wikipedia , lookup
Computational phylogenetics wikipedia , lookup
Smith–Waterman algorithm wikipedia , lookup
Sequence alignment wikipedia , lookup
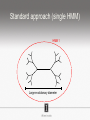
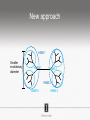
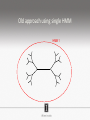
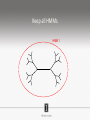
Ensembles of HMMs and their use in biomolecular sequence analysis Nam-phuong Nguyen Carl R. Woese Institute for Genomic Biology University of Illinois at Urbana-Champaign Human Microbiome • 10 times more bacteria cells than human cells • Important role in regulating health • Disruption associated with risk factors for diseases Metagenomics • Analyzing DNA sequences from environmental sample • Sequencing technology produces short fragments of DNA • Typical datasets contain millions of reads Phylogenetic pipeline Hu Ch Go Or = = = = AGGCTATCACCGACTCCA TAGCTATCACGACCGC TAGCTGACCGC TCACGACCGACA Hu Ch Go Or = = = = -AGGCTATCACGACCTCCA TAG-CTATCACGACCGC-TAG-CT-----GACCGC--------TCACGACCGACA Using the MSA and tree to identify reads Hu Ch Go Or Qu = = = = = -AGGCTATCACGACCTCCA TAG-CTATCACGACCGC-TAG-CT-----GACCGC--------TCACGACCGACA TCACCCC -------TCACC-CC---- Q Represent MSA using a profile Hidden Markov Model (HMM) Phylogenetic Placement • Align each query sequence to backbone alignment: • HMMALIGN (Eddy, Bioinformatics 1998) • PaPaRa (Berger and Stamatakis, Bioinformatics 2011) • Place each query sequence into backbone tree, using extended alignment: • pplacer (Matsen et al., BMC Bioinformatics 2010) • EPA (Berger et al., Systematic Biology 2011) Phylogenetic Placement • Align each query sequence to backbone alignment: • HMMALIGN (Eddy, Bioinformatics 1998) • PaPaRa (Berger and Stamatakis, Bioinformatics 2011) • Place each query sequence into backbone tree, using extended alignment: • pplacer (Matsen et al., BMC Bioinformatics 2010) • EPA (Berger et al., Systematic Biology 2011) HMMER and PaPaRa results Backbone size: 500 5000 fragments 20 replicates 0.0 Increasing rate evolution Standard approach (single HMM) HMM 1 Large evolutionary diameter New approach HMM 1 Smaller evolutionary diameter HMM 1 HMM 2 HMM 2 HMM 3 HMM 4 Ensemble of HHMs (eHMMs) HMM 1 HMM 3 HMM 2 HMM 4 SEPP (10% rule) Simulated Results Backbone size: 500 5000 fragments 20 replicates 0.0 Increasing rate evolution Summary so far • Use DNA sequences to build an MSA and tree • Use an existing MSA and tree to identify a sequence • eHMMs for aligning a sequence to an existing MSA Metagenomic taxon identification Objective: classify short reads in a metagenomic sample Abundance profiling Objective: distribution of the species (or genera, or families, etc.) within the sample For example, the distribution of a sample at the species level might be: Species A: 10% Species B: 25% Species C: 55% Species D: 1% Species E: 9% Genome-based profiling A A B Population of 2 bacteria, A and B. B has twice as large genome as A. True profile: 67% A, 33% B Profile estimated from reads: 50% A, 50%B Single copy marker-based profiling A A Population of 2 bacteria, A and B. B has twice as large genome as A. Each have a single copy of gene C B True profile: 67% A, 33% B Profile estimated from reads: 67% A, 33%B TIPP: Taxonomic Identification and Phylogenetic Profiling Fragmentary unknown reads for a gene Known full length sequences for a gene, and an alignment and a tree ACCG CGAG CGG GGCT … … … … ACCT AGG...GCAT (species1) TAGC...CCA (species2) TAGA...CTT (species3) AGC...ACA (species4) ACT..TAGAA (species5) TIPP: Taxonomic Identification and Phylogenetic Profiling • Nguyen et al., Bioinformatics, 2014 Reads Assign to marker genes Marker genes Classify reads Compute profile Abundance profiling • Objective: Distribution of the species (or genera, or families, etc.) within the sample. • Leading techniques: – PhymmBL (Brady & Salzberg, Nature Methods 2009) – NBC (Rosen, Reichenberger, and Rosenfeld, Bioinformatics 2011) – MetaPhyler (Liu et al., BMC Genomics 2011), from the Pop lab at the University of Maryland – MetaPhlAn (Segata et al., Nature Methods 2012), from the Huttenhower Lab at Harvard – mOTU (Bork et al., Nature Methods 2013) • MetaPhyler, MetaPhlAn, and mOTU are marker-based techniques (but use different marker genes). “Hard” genome datasets (known genomes and high indel error) Note: NBC, MetaPhlAn, and Metaphyler cannot classify any sequences from at least of the high indel long sequence datasets. mOTU terminates with an error message on all the high indel datasets. “Novel” genome datasets Note: mOTU terminates with an error message on the long fragment datasets and high indel datasets. TIPP compared to other profiling methods • TIPP is highly accurate, even in the presence of novel genomes and high sequencing error • All other methods are less robust • Accurate profiles can be estimated using only a portion of the reads Ensemble of HMMs • Represent MSA using many HMMs • Modifications enable – Fast and accurate alignment of fragmentary and ultra-large datasets (Nguyen et al., Genome Biology 2015 ) – Improved protein homology detection (in preparation) • Currently in use for – – – – – Vertebrate nuclear receptor evolution (in preparation) 1KP Plant phylogenomics study (in preparation) Identification of cardioviruses in rats (in preparation) Identification of microbial sample (in preparation) and many others… Ensemble of HMMs • Represent MSA using many HMMs • Modifications enable – Fast and accurate alignment of fragmentary and ultra-large datasets (Nguyen et al., Genome Biology 2015 ) – Improved protein homology detection (in preparation) • Currently in use for – – – – – Vertebrate nuclear receptor evolution (in preparation) 1KP Plant phylogenomics study (in preparation) Identification of cardioviruses in rats (in preparation) Identification of microbial sample (in preparation) and many others… Real biological data is messy Full-length P450 gene ~500 amino acid residues Total sequences before filtering ~225K Challenge: How do we align large datasets with fragmentary sequences? HMMs for MSA Given seed alignment and a collection of sequences for the protein family: Represent seed alignment using a profile HMM Align each additional sequence to the HMM Use transitivity to obtain MSA Drawbacks: Requires seed alignment Poor accuracy on evolutionarily divergent datasets Old approach using single HMM HMM 1 SEPP/TIPP approach HMM 1 HMM 3 HMM 2 HMM 4 How small of a subset size do we go to? HMM 1 HMM 3 HMM 2 HMM 4 Keep all HMMs HMM 1 Keep all HMMs HMM 1 HMM 2 HMM 3 Nested Hierarchical Ensemble of HMMs HMM 1 HMM 2 HMM 4 m HMM 7 HMM 3 HMM 5 HMM 6 UPP: Ultra-large alignment using phylogeny aware profiles UPP: Ultra-large alignment using phylogeny aware profiles UPP: Ultra-large alignment using phylogeny aware profiles UPP: Ultra-large alignment using phylogeny aware profiles UPP: Ultra-large alignment using phylogeny aware profiles UPP: Ultra-large alignment using phylogeny aware profiles Experimental Design Examined both simulated and biological DNA, RNA, and AA datasets Generated fragmentary datasets from the full-length datasets Explored impact of algorithmic design Compared Clustal-Omega, Mafft, Muscle, PASTA, and UPP ML trees estimated on alignments Scored alignment and tree error 5/14/14 Alignment error measured as average of SPFN and SPFP Tree error measured in FN rate or Delta FN rate UPP Algorithmic Parameters • Decompose or not? Use an ensemble of HMMs or just a single HMM? • Use a small (100 sequence) or large (1000 sequence) backbone? RNASim Alignment Error Full-length datasets 5/14/14 Alignment error on fragmentary 16S.T Fragmentary datasets 5/14/14 Running time on simulated RNA datasets UPP has close to linear runtime scaling UPP compared to other alignment methods • PASTA and UPP result in accurate alignments and trees on full-length sequences (PASTA slightly more accurate trees) • UPP is more robust on fragmentary data • Using combination of UPP+PASTA can give best overall result Summary • Ensemble of HMMs • TIPP for identification and profiling • UPP for ultra-large alignment Acknowledgements • Illinois • Tandy Warnow • Rebecca Stumpf • Bryan White • Mike Nute • Brenda Wilson • UCSD • Siavash Mirarab • UMD • Mihai Pop • Bo Liu • U of Copenhagen • Alonzo Alfaro-Núñez • Tom Hansen • Anders Hansen • Funding • NSF 09-35347 • NSF 08-20709 • NSF 0733029 • University of Alberta Questions? • Available at https://github.com/smirarab/sepp