Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Two-hybrid screening wikipedia , lookup
Transcriptional regulation wikipedia , lookup
Lipid signaling wikipedia , lookup
Point mutation wikipedia , lookup
Silencer (genetics) wikipedia , lookup
Catalytic triad wikipedia , lookup
Biochemical cascade wikipedia , lookup
Proteolysis wikipedia , lookup
Paracrine signalling wikipedia , lookup
Oxidative phosphorylation wikipedia , lookup
Siderophore wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Human iron metabolism wikipedia , lookup
Biosynthesis wikipedia , lookup
Evolution of metal ions in biological systems wikipedia , lookup
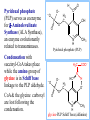
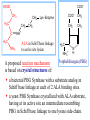
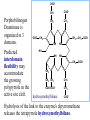
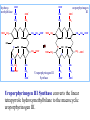
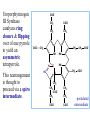
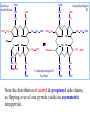
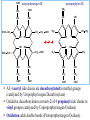
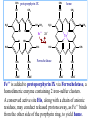
Molecular Biochemistry II Synthesis of Heme Copyright © 1999-2008 by Joyce J. Diwan. All rights reserved. CH3 CH3 S HC CH2 protein N H3C CH3 N OOC CH2 CH2 Fe N CH N S CH2 protein CH3 CH2 CH3 CH2 COO Heme c Heme is the prosthetic group of hemoglobin, myoglobin, & cytochromes. Heme is an asymmetric molecule. E.g., note the positions of methyl side chains around the ring system. The heme ring system is synthesized from glycine & succinyl-CoA. Using isotopic tracers, it was initially found that N & C atoms of heme are derived from glycine and acetate. It was later determined that the labeled acetate enters Krebs Cycle as acetyl-CoA, and the labeled carbon becomes incorporated into succinyl-CoA, the more immediate precursor of heme. O OOC CH2 CH2 C S-CoA + succinyl-CoA d-Aminolevulinic Acid Synthase OOC CH2 NH3+ glycine H+ CoA-SH CO2 O OOC CH2 CH2 C CH2 NH3+ d-aminolevulinate (ALA) Heme synthesis begins with condensation of glycine & succinyl-CoA, with decarboxylation, to form d-aminolevulinic acid (ALA). H Pyridoxal phosphate (PLP) serves as coenzyme for d-Aminolevulinate Synthase (ALA Synthase), an enzyme evolutionarily related to transaminases. Condensation with succinyl-CoA takes place while the amino group of glycine is in Schiff base linkage to the PLP aldehyde. CoA & the glycine carboxyl are lost following the condensation. O O P O C H2 C OH O O N CH3 H Pyridoxal phosphate (PLP) H2C COO N+ HC O O H2 C P O H O O N H CH3 glycine-PLP Schiff base (aldimine) ALA Synthase is the committed step of the heme synthesis pathway, & is usually rate-limiting for the overall pathway. Regulation occurs through control of gene transcription. Heme functions as a feedback inhibitor, repressing transcription of the ALA Synthase gene in most cells. A variant of ALA Synthase expressed only in developing erythrocytes is regulated instead by availability of iron in the form of iron-sulfur clusters. COO COO CH2 CH2 CH2 CH2 C + O C PBG Synthase 2 H2O O CH2 CH2 NH3+ NH3+ 2 d-aminolevulinate (ALA) H2C COO COO CH2 CH2 CH2 C C C CH NH3+ N H porphobilinogen (PBG) PBG Synthase (Porphobilinogen Synthase), also called ALA Dehydratase, catalyzes condensation of two molecules of d-aminolevulinate to form the pyrrole ring of porphobilinogen (PBG). The reaction mechanism involves 2 Lys residues & a bound cation at the active site. Cation in mammalian enzyme is Zn++. HOOC CH2 H2C CH2 LysEnzyme CH2 CH2 C H2C NH2 NH+ ALA in Schiff base linkage to active site lysine As each of the two d-aminolevulinate (ALA) substrates binds at the active site, its keto group initially reacts with the side-chain amino group of one of the two lysine residues to form a Schiff base. These Schiff base linkages promote the C-C and C-N condensation reactions that follow, assisted by the metal ion that coordinates to the ALA amino groups. COO HOOC CH2 H2C CH2 LysEnzyme CH2 CH2 C H2C NH2 COO CH2 CH2 CH2 NH+ ALA in Schiff base linkage to active site lysine A proposed reaction mechanism is based on crystal structures of: H2C NH3 + N H Porphobilinogen (PBG) a bacterial PBG Synthase with a substrate analog in Schiff base linkage at each of 2 ALA binding sites. a yeast PBG Synthase crystallized with ALA substrate, having at its active site an intermediate resembling PBG in Schiff base linkage to one lysine side-chain. The Zn++ binding sites in the homo-octomeric mammalian Porphobilinogen Synthase, which include cysteine S ligands, can also bind Pb++ (lead). Inhibition of Porphobilinogen Synthase by Pb++ results in elevated blood ALA, as impaired heme synthesis leads to de-repression of transcription of the ALA Synthase gene. High ALA is thought to cause some of the neurological effects of lead poisoning, although Pb++ COO COO also may directly affect the nervous system. ALA is toxic to the brain, perhaps due to: • Similar ALA & neurotransmitter GABA (g-aminobutyric acid) structures. • ALA autoxidation generates reactive oxygen species (oxygen radicals). CH2 CH2 CH2 CH2 C O CH2 CH2 NH3+ NH3+ ALA GABA COO N H pyrrole Porphobilinogen (PBG) is the first pathway intermediate that includes a pyrrole ring. COO CH2 CH2 CH2 H2C NH3 + N H Porphobilinogen (PBG) The porphyrin ring is formed by condensation of 4 molecules of porphobilinogen. Porphobilinogen Deaminase catalyzes successive PBG condensations, initiated in each case by elimination of the amino group. COO- Enz S COO COO- CH2 COO- CH2 CH2 CH2 CH2 CH2 N H dipyrromethane - N H PBG Deaminase PDB 1PDA Porphobilinogen Deaminase has a dipyrromethane prosthetic group, linked at the active site via a cysteine S. The enzyme itself catalyzes formation of this prosthetic group. COOCOO- CH2 COO- COO-CH COO- Enz S COO- CH2 COO- CH2 CH2 CH2 N H CH2 CH2 2 CH2 CH2 CH2 N H NH HN NH HN CH2 COO- CH2 COOCH2 CH2 CH2 CH2 COO-COO- CH2 COO- PBG units are added to the dipyrromethane until a linear hexapyrrole has been formed. COO- Porphobilinogen Deaminase is organized in 3 domains. Predicted interdomain flexibility may accommodate the growing polypyrrole in the active site cleft. - OOC CH2 COO- CH2 CH2 CH2 NH HN NH HN CH2 CH2 COO- CH2 COO- HO CH2 CH2 CH2 CH2 COO-COO- CH2 hydroxymethylbilane COO- Hydrolysis of the link to the enzyme's dipyrromethane releases the tetrapyrrole hydroxymethylbilane. COO- hydroxymethylbilane - OOC COO- uroporphyrinogen III - CH2 COO- CH2 COO CH2 CH2 CH2 CH2 CH2 CH2 NH HN NH HN CH2 COO- - OOC CH2 NH HN NH HN CH2 CH2 COO- CH2 COO- HO C C CH2 C COO- - OOC CH2 C CH2 CH2 CH2 CH2 COO-COO- CH2 COO- Uroporphyrinogen III Synthase CH2 CH2 CH2 CH2 COO- COO- Uroporphyrinogen III Synthase converts the linear tetrapyrrole hydroxymethylbilane to the macrocyclic uroporphyrinogen III. Uroporphyrinogen III Synthase catalyzes ring closure & flipping over of one pyrrole to yield an asymmetric tetrapyrrole. This rearrangement is thought to proceed via a spiro intermediate. COO- - OOC CH2 COO- CH2 CH2 CH2 CH2 N NH HN C HN CH2 HC CH2 COO- COO- CH2 CH2 COOCH2 CH2 COO- COO- CH2 postulated intermediate COO- hydroxymethylbilane - OOC COO- uroporphyrinogen III - CH2 COO- CH2 COO CH2 CH2 CH2 CH2 CH2 CH2 NH HN NH HN CH2 COO- - OOC CH2 NH HN NH HN CH2 CH2 COO- CH2 COO- HO C C CH2 C - COO - OOC CH2 C CH2 CH2 CH2 CH2 COO-COO- CH2 COO- Uroporphyrinogen III Synthase CH2 CH2 CH2 CH2 COO- COO- Note the distribution of acetyl & propionyl side chains, as flipping over of one pyrrole yields an asymmetric tetrapyrrole. The active site of Uroporphyrinogen III Synthase is in a cleft between two domains of the enzyme. The structural flexibility inherent in this arrangement is proposed to be essential to catalysis. Uroporphyrinogen III PDB 1JR2 Synthase Uroporphyrinogen III is the precursor for synthesis of vitamin B12, chlorophyll, and heme, in organisms that produce these compounds. Conversion of uroporphyrinogen III to protoporphyrin IX occurs in several steps. - COO - protoporphyrin IX uroporphyrinogen III CH2 COO- CH2 CH2 CH2 CH OOC CH2 CH2 - CH2 COO CH3 CH CH2 H3C NH HN NH NH HN N - CH2 OOC CH2 COO- N HN H3C CH3 CH2 CH2 CH2 CH2 CH2 CH2 CH2 CH2 COO- COO- COO- COO- All 4 acetyl side chains are decarboxylated to methyl groups (catalyzed by Uroporphyrinogen Decarboxylase) Oxidative decarboxylation converts 2 of 4 propionyl side chains to vinyl groups (catalyzed by Coproporphyrinogen Oxidase) Oxidation adds double bonds (Protoporphyrinogen Oxidase). CH2 protoporphyrin IX CH2 CH CH3 CH CH CH2 H3C NH N N Fe++ heme CH3 CH CH2 H3C Fe HN N H3C CH3 CH2 CH2 CH2 COO- N N 2H+ N H3C Ferrochelatase CH3 CH2 CH2 CH2 CH2 CH2 COO- COO- COO- Fe++ is added to protoporphyrin IX via Ferrocheletase, a homodimeric enzyme containing 2 iron-sulfur clusters. A conserved active site His, along with a chain of anionic residues, may conduct released protons away, as Fe++ binds from the other side of the porphyrin ring, to yield heme. Regulation of transcription or post-translational processing of enzymes of the heme synthesis pathways differs between erythrocyte forming cells & other tissues. In erythrocyte-forming cells there is steady production of pathway enzymes, limited only by iron availability. In other tissues expression of pathway enzymes is more variable & subject to feedback inhibition by heme. Porphyrias are genetic diseases in which activity of one of the enzymes involved in heme synthesis is decreased (e.g., PBG Synthase, Porphobilinogen Deaminase, etc…). Symptoms vary depending on the enzyme the severity of the deficiency whether heme synthesis is affected primarily in liver or in developing erythrocytes. Occasional episodes of severe neurological symptoms are associated with some porphyrias. Permanent nerve damage and even death can result, if not treated promptly. Elevated d-aminolevulinic acid (ALA), arising from de-repression of ALA Synthase gene transcription, is considered responsible for the neurological symptoms. Photosensitivity is another common symptom. Skin damage may result from exposure to light. This is attributable to elevated levels of light-absorbing pathway intermediates and their degradation products. Question: How do you think episodes of acute neurological symptoms would be treated? Treatment is by injection of hemin (a form of heme). Why would this work? The heme, in addition to supplying needs, would repress transcription of the gene for ALA Synthase, rate-limiting for the pathway and the source of excess ALA. View an amination of the heme synthesis pathway. Regulation of iron absorption & transport. Iron for synthesis of heme, Fe-S centers & other non-heme iron proteins is obtained from: the diet release of recycled iron from macrophages of the reticuloendothelial system that ingest old & damaged erythrocytes. There is no mechanism for iron excretion. Iron is significantly lost only by bleeding, including menstruation in females. Small losses occur from sloughing of cells of skin & other epithelia. transferrin with bound Fe extracellular space transferrin receptor receptor-mediated endocytosis Iron is transported in blood serum bound to the protein transferrin. The plasma membrane transferrin receptor mediates uptake of the complex of iron with transferrin by cells via receptor mediated endocytosis. Iron is stored within cells as a complex with the protein ferritin. The main storage site is liver. The plasma membrane protein ferroportin mediates: release of absorbed iron from intestinal cells to blood serum release of iron from hepatocytes (liver cells) and macrophages. Control of dietary iron absorption and serum iron levels involves regulation of ferroportin expression. Transcription of the gene for the iron transporter ferroportin is responsive to iron. Hepcidin, a regulatory peptide secreted by liver, induces degradation of ferroportin. Hepcidin secretion increases when iron levels are high or in response to cytokines produced at sites of inflammation. Degradation of ferroportin leads to decreased absorption of dietary iron and decreased serum iron. Hepcidin is considered an antimicrobial peptide because by lowering serum iron it would limit bacterial growth. Hereditary hemochromatosis is a family of genetic diseases characterized by excessive iron absorption, transport & storage. Genes mutated in these disorders include those for: transferrin receptor a protein HFE that interacts with transferrin receptor hepcidin hemojuvelin, an iron-sensing protein required for transcription of the gene for hepcidin. E.g., impaired synthesis or activity of hepcidin leads to unrestrained ferroportin activity, with high dietary intake and high % saturation of serum transferrin with iron. Organs particularly affected by accumulation of excess iron include liver and heart.