Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
List of types of proteins wikipedia , lookup
Protein design wikipedia , lookup
Protein domain wikipedia , lookup
Circular dichroism wikipedia , lookup
Protein folding wikipedia , lookup
Bimolecular fluorescence complementation wikipedia , lookup
Homology modeling wikipedia , lookup
Intrinsically disordered proteins wikipedia , lookup
Degradomics wikipedia , lookup
Protein structure prediction wikipedia , lookup
Protein moonlighting wikipedia , lookup
Protein purification wikipedia , lookup
Western blot wikipedia , lookup
Nuclear magnetic resonance spectroscopy of proteins wikipedia , lookup
Protein–protein interaction wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
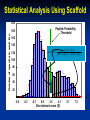
Analysis of Complex Proteomic Datasets Using Scaffold Free Scaffold Viewer can be downloaded at: www.proteomesoftware.com Scaffold: Why do we need it? Shotgun proteomics Analysis of complex mixtures Whole cell extract 10,000+ proteins 600,000 peptides 1.2 Million Spectra!!! • Beyond the realm of manual interpretation • How do we determine what is a valid protein identification? Statistical Analysis Using Scaffold • All search engines use different scoring algorithms Can not directly compare results • Many search engines results are described by more than one value Examples: Mascot Ion Score and Identity Score Sequest Xcorr and DeltaCn Statistical Analysis Using Scaffold Peptide Prophet* • Creates a universal score (discriminant score) for the search engine result (e.g. XCorr and DeltaCn are compressed to one score for SEQUEST results, Ion score and Identity score for Mascot results) • Plots a histogram of the discriminant scores and calculates a bimodal distribution based on standard statistics to differentiate between correct and incorrect hits • Computes the probability that the match is correct at a given discriminant score *Nesvizhskii, A. I. et al, Anal. Chem. 2003, 75, 4646-4658 Statistical Analysis Using Scaffold 200 Number of spectra in each bin 180 Histogram of discriminate scores 160 140 120 100 80 60 40 20 0 -3.9 -2.3 -0.7 0.9 2.5 4.1 Discriminant score (D) 5.7 7.3 Statistical Analysis Using Scaffold 200 Number of spectra in each bin 180 160 Assumes a mixture of standard statistical distributions “incorrect” 140 120 100 “correct” 80 60 40 20 0 -3.9 -2.3 -0.7 0.9 2.5 4.1 Discriminant score (D) 5.7 7.3 Statistical Analysis Using Scaffold 200 Number of spectra in each bin 180 Peptide Probability Threshold “incorrect” 160 140 p ( D | ) p ( ) p( D | ) p() p( D | ) p( ) “correct” p ( | D) 120 100 80 60 40 20 0 -3.9 -2.3 -0.7 0.9 2.5 4.1 Discriminant score (D) 5.7 7.3 Statistical Analysis Using Scaffold One Search Engine may not be enough SEQUEST 9% 22% 4% 34% X!Tandem www.proteomesoftware.com 19% 7% 5% Mascot Statistical Analysis Using Scaffold • Peptide Prophet statistics are applied separately for each search engine result (i.e. Mascot, SEQUEST, and X!Tandem) • Scaffold Merger combines the peptide probabilities from each search engine to generate a protein probability The probability of identifying a spectrum + The probability of agreement between search engines Protein Probability Statistical Analysis Using Scaffold Advantages using of Scaffold • Allows you to choose a statistical error rate by setting probability thresholds • Allows you to compare and combine results from different experiments and different search engines • Allows sharing of raw data and search results • Accepted as a suitable statistical method to validate large datasets This is the Samples view List of all the proteins found in your samples Homologous proteins (proteins matched to the same peptides) are shown. You can directly like out to database entries How does Scaffold Deal with peptides that can be assigned to more than one protein? General Rule Explain the spectral data with the smallest set of proteins B A Protein A and Protein B share all the same peptides so they will be grouped together How does Scaffold Deal with peptides that can be assigned to more than one protein? General Rule Explain the spectral data with the smallest set of proteins B A Protein A and protein B each have one unique peptide they will be listed separately only if the peptide probability is > 50% How does Scaffold Deal with peptides that can be assigned to more than one protein? General Rule Explain the spectral data with the smallest set of proteins B A Protein B has two unique peptides it will be listed separately Scaffold will extract GO terms from NCBI annotations Gene Ontology “GO” terms • Controlled vocabulary containing consistent descriptions of gene products in different databases • Describe gene products in terms of their associated biological processes, cellular components and molecular functions in a species independent manner Gene Ontology Project http://www.geneontology.org/GO.doc.shtml List of samples Probability thresholds for peptide and protein identifications and required number of unique peptides can be defined Color coded to represent probability that protein identification is correct This is the Proteins view Spectrum of each peptide labeled with y and b ions which can be used for manual validation Manual Spectrum Evaluation • Search engine scores Is peptide found by more than one search engine? Mascot ion score > 40 SEQUEST Xcorr > 2 (+2 ion), 2.5 (+3 ion) deltaCn > 0.2 • Good signal-to-noise • Long stretches of y and/or b ions • All dominant peaks are assigned as y or b ions • Fragmentation chemistry N-terminal cleavage at P dominate y-ion C-terminal cleavage at D and E dominate b-ion Peptides containing W abundant y-ions S and T tend to lose water (-18 Da) R, N, and Q tend to lose ammonia (-17 Da) Good Spectrum Peptide Sequence IAELAGFSVPENTK +2 charge on parent peptide y5 100% I A K L E T N A E G P F V S S V F 1474.73 AMU, +2 H (Parent Error: -650 ppm) P G E A N L T E K A I Good signal-to-noise 50% b9-H2O y6 b7 b5 b6 b3 y3 b4 y4 0% 0 250 500 y7 b8 b9 y9 y10 y8 b10 750 m/z 1000 b11y11 b12 b13 y12 1250 Good coverage of y and b ion series Dominant y-ion at N-terminal cleavage of P SEQUEST: Xcorr = 2.61 deltaCn = 0.4 Mascot: Ion Score = 60.1 Identify Score = 37.3 Bad Spectrum Peptide Sequence YPLADYALTPDMAIVDANLVMDMPK +3 charge on parent peptide 100% Y K P P L M A D D Y M V b19+2H b20+2H b21+2H+1 A L T P D M L N A D V I internal PLADYALTPD-CO b9 A A I M V D 2767.75 AMU, +3 H (Parent Error: -240 ppm) D A N L V M D M P K P T L A Y D A L P Y Poor coverage of y and b ion series b17-H2O+2H b8 b9+1 b20+2H+1 b9-H2O-H2O+2H b17+2H x17 y15+2H b8-H2O b13+2H+1 y4 y3 x8+2H a7-H2O+1 b9+2 y10 b22+2H+1 internalb13+2H PLADYAL-NH3 b9-H2O-H2O 50% b5y5 y6 y7 y9 y11 b11 y12 b15 b14 0% 0 500 1000 1500 2000 2500 m/z Multiple unassigned peaks SEQUEST: Xcorr = 2.26 deltaCn = 0.2 Poor signal-to-noise Mascot: Ion Score = 9.93 Identity Score = 37.3 This is the Statistics view Scaffold Statistics View Score Histogram Blue indicates “incorrect” proteins Red indicates “correct” proteins Important! Must have enough data to fit two distributions for the statistics to be valid. Protein is “correct” if it passes the peptide and protein probability and minimum # peptide filters. Scaffold Statistics View With at least 2 unique Peptides (95% peptide prob) the maximum protein probability is ~100%. With only 1 unique peptide (95% peptide prob) the maximum protein probability is <90%. Scaffold Statistics View Missed IDs SEQUEST only Scaffold Statistics View Mascot only Missed IDs Scaffold Statistics View Using both Mascot and Sequest results in more “correct” protein identifications Mascot only Both Sequest only This is the Publish View Publication Guidelines for Proteomic Data Journal of Molecular and Cellular Proteomics http://www.mcponline.org/misc/ParisReport_Final.shtml Publication Guidelines for Proteomic Data Data Analysis • Name and version of software used to extract peak list • Name and version of database searching software (Mascot, Sequest, Spectrum Mill, or X! Tandem) • Values of all search parameters used (enzyme, modifications, mass tolerance, etc.) • Name and size of the database searched (Swisprot or NCBI and the number of sequence entries) • Name and version of any additional software used for statistical analysis and an explanation of the analysis (Scaffold, #peptide requirements, probability settings) Publication Guidelines for Proteomic Data Each Peptide Identified • Peptide sequence noting any modifications or missed cleavages • Parent peptide ion mass and charge • All search engine scores Each Protein Identified • Accession number • Sequence coverage and total number of unique peptides