Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Folding@home wikipedia , lookup
Rosetta@home wikipedia , lookup
Protein design wikipedia , lookup
Bimolecular fluorescence complementation wikipedia , lookup
Circular dichroism wikipedia , lookup
Protein moonlighting wikipedia , lookup
List of types of proteins wikipedia , lookup
Western blot wikipedia , lookup
Structural alignment wikipedia , lookup
Protein purification wikipedia , lookup
Intrinsically disordered proteins wikipedia , lookup
Alpha helix wikipedia , lookup
Protein mass spectrometry wikipedia , lookup
Homology modeling wikipedia , lookup
Protein folding wikipedia , lookup
Protein domain wikipedia , lookup
Protein–protein interaction wikipedia , lookup
Nuclear magnetic resonance spectroscopy of proteins wikipedia , lookup
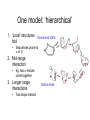
Last Tuesday and Beyond • Common 2° structural elements: influenced by 1° structure – – – • alpha helices beta strands beta turns Structure vs. function – – • Fibrous (collagen, silk) Globular Determination of 3-D structure – – X-ray crystallography NMR Organization of protein structure • Subdomains within proteins • Motifs: common ways 2° structural elements interact • The process of protein folding and unfolding 3-D substructure • Domains – Compact, globular units found in large proteins – Folds independently – Retain structure even when separated from rest of protein – Genetic manipulation: » Can be added, removed, swapped…(often) with impunity – Result from gene fusion during evolution – Often bring together different functions » Regulatory & catalytic Myosin “Supersecondary” structures Common, stable motifs in which 2o structural elements come together • • • • • • • b-a-b loop a-a corner b-barrels Greek key Jellyroll b-meander a-a unit Greek key b-meander Jellyroll Rules for protein folds 1. Burial of H-phobic – Requires 2 layers of 2° structure 2. a-helix and b-sheets – Different ‘layers’ of structure because Hbonding 3. Adjacent peptide segments in structure, sometimes adjacent in sequence 4. Connections cannot cross/form knots 5. b-conformation most stable with right twist Simplifying 3D structure • Classify according to 2° components • • • • All a All b a/b (alternate) a & b (segregated) • Many different folds • But fewer than 1000 may exist in all proteins • 3o structure conserved 3-D structure examples Family and superfamily Multisubunit proteins • Separate subunits – Sometimes different domains and functions • Catalysis/regulation • Structural/catalysis • Multistep catalysis • Hemoglobin – – – – – 4 polypeptide chains 4 prosthetic (heme) groups 2 a chains (141 AA) 2 b chains (146 AA) Arranged as symmetric pairs • a/b subunit • Tetramer or dimer of a/b protomers Limitations on protein size – Theoretically unlimited, but not so in practice – Genetic coding capacity of nucleic acids – Accuracy of protein biosynthesis • More efficient to make many copies of small than one large protein • >~100000: multiple subunits (more than one polypeptide chain, more than one gene) – Reduce probability for error • 1/10000 amino acid Protein folding • Ribonuclease – ‘Denatured’ with urea – Disulfides broken with a ‘reducing agent’ (BME) • inactive protein – Urea and BME removed • Active, refolded protein • Protein has ‘renatured’ – Sequence confers 3-D structure activity Protein denaturation • 1. 2. 3. 4. 5. 6. 7. 8. Denaturants Heat pH (strong acids/bases) Organic solvents Salts (urea, guanidine HCl) Detergents Reducing agents Heavy metal ions Mechanical stress • Only ‘weak’ and S-S interactions are broken – No peptide covalent bonds • Detect by spectroscopy, eg. fluorescence of aromatic residues Protein folding: strand → native • Cannot be completely random – – – – – 100 residues: 10100 possible conformations Theoretically 1077 years for protein to fold Actual time scale: milliseconds to seconds “Levinthal’s paradox” How? • Driven by physics/chemistry: Anfinsen’s experiment One model: ‘hierarchical’ 1. ‘Local’ structures fold • Denatured state Sequences prone to a or b 2. Mid-range interaction • eg. two a helices come together 3. Longer range interactions • Two loops interact Native state “Molten globule” model • Molten globule model – Peptide collapses into compact state • • – • Hphobic on inside, Hphilic on outside ‘molten globule’ Protein folds from this Describe with a free energy funnel – Thousands of unfolded conformations • – Some collapse, some start forming 2o structure • – Highly unstable Semistable intermediates At bottom, single/few native structures with a small set of conformation