Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Rosetta@home wikipedia , lookup
Protein design wikipedia , lookup
Homology modeling wikipedia , lookup
Protein domain wikipedia , lookup
Circular dichroism wikipedia , lookup
Protein folding wikipedia , lookup
Protein structure prediction wikipedia , lookup
Protein moonlighting wikipedia , lookup
Bimolecular fluorescence complementation wikipedia , lookup
List of types of proteins wikipedia , lookup
Intrinsically disordered proteins wikipedia , lookup
Nuclear magnetic resonance spectroscopy of proteins wikipedia , lookup
Degradomics wikipedia , lookup
Protein purification wikipedia , lookup
Western blot wikipedia , lookup
Protein–protein interaction wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
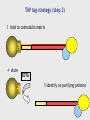
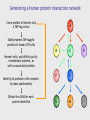
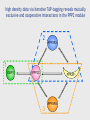
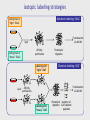
MBP1001 Advanced Cell Biology 2010 Proteomics and Mass Spectrometry Brian Raught [email protected] Proteomics is an extremely powerful and broadly applicable technology can be used to identify e.g. low stoichiometry PTMs, components of protein complexes, or to characterize all protein components in an organelle, tissue or organism the key - but poorly understood - technology in this process is mass spectrometry-based peptide sequencing today’s lecture will provide a brief overview of the approach, followed by some examples of its utility First step- sample preparation the goal - simplify depending upon the goal of your experiment, you will isolate large or small numbers of proteins for analysis you may subject your protein population to one or more fractionation steps, e.g. 1D SDS-PAGE 2D gel electrophoresis strong cation exchange liquid chromatography newer technologies - free flow electrophoresis you will then convert your protein sample to peptides Why are peptides (and not proteins) sequenced? top-down approaches can identify intact proteins, but... proteins can be difficult to handle, and all proteins in your sample may not be soluble under the same conditions (e.g. membrane-spanning proteins vs DNA binding prots) proteins are often significantly processed and modified, resulting in many different isoforms, making identification difficult ion trap mass spectrometers are most efficient at obtaining sequence info from peptides up to ~40aa in length – ID of prots via peptides is bottom-up proteomics Proteases are used to convert proteins to peptides trypsin stable and very active, cleaves on the carboxy-terminal side of K and R residues (except when modified or followed by a P) results in information rich, easily interpretable peptide fragment spectra other commonly used proteases LysC AspN GluC sequence non-specific proteases are generally avoided, since they divide the peptide signal into multiple overlapping species, and thereby generate unnecessarily complex peptide mixtures How are peptides introduced into the mass spectrometer? 1. liquid chromatography (LC) directly coupled (in-line) with MS (LC-MS), introduced via electrospray (ESI) 2. peptides spotted onto metal surface, released into the MS via controlled laser shots (MALDI) LC-MS peptides are loaded onto an extremely small (50-150um) reversed-phase (silica particles coated with C18) column, and eluted directly into the machine by a gradient of increasing organic solvent (water - acetonitrile, with a small amount of acid – pH~2) 100-400nl/min flow rates (nanoflow) separated according to hydrophobicity (standard 1-2hr runs) eluted into the MS in a very small volume, and therefore at high concentrations In most MS applications, peptides are positively charged, via the application of a strong current to the buffer in the LC column (~2kV) some amino acids, as well as the peptide amino terminus, are positively charged at low pH (e.g. K, R, H) – so most peptides (esp. tryptic peptides) are multiply charged charge is critical - the MS optics manipulate only charged ions, whereas uncharged peptides are “invisible” LC column ends in a very fine needle (~5microns); since the HPLC system is under pressure, and an electrical charge is applied, this results in a fine spray of droplets emanating from the tip containing charged peptides – electrospray ionization (soft ionization = Nobel prize) Positively charged peptides are guided into the machine by a strong charge potential (and vacuum) peptides first enter a small heated tube - as the fine droplets containing the peptides traverse the length of the tube, the buffer is rapidly evaporated as the concentration of positively charged peptides increases in smaller and smaller droplets, they begin to repel one another, resulting in a series of Coulombic explosions end result - individual positively charged peptides in the gas phase are ready for manipulation and measurement So what is in a mass spectrometer, anyway? think of it as a series of boxes, connected to each other via a pipe - each box has the ability to trap and release peptides, some boxes can also smash your peptides at the end of the pipe sits a peptide counter (detector) 1 2 3 detector Step 1 peptides enter the first chamber (Q1), where they are trapped (until the trap is full) typical ion traps (Paul trap) use a combination of static DC and RF oscillating AC electric fields to move and manipulate the charged molecules to characterize the contents of the trap, a small amount of the peptides (~10%) is released to the detector this process is called the parent ion, precursor, or MS scan, and yields the m/z and intensity of all of the peptides in the first chamber at that moment readout is expressed as intensity of signal (number of counts) for a given mass (actually m/z or mass/charge) a parent ion (MS) scan ion intensity select for fragmentation m/z Step 2 collision induced dissociation a process whereby a (mostly) pure population of a single peptide (actually a small m/z window) is ejected to a second chamber (the collision cell), and mixed with an inert gas as energy is applied to the isolated peptide population, they collide with the gas particles, and fragment – luckily for us, most of the time peptides fragment at peptide (amide) bonds between amino acids add just enough energy to the collision cell such that an individual peptide fragments just once the resulting mixed populations of peptide fragments is then analyzed to give a product ion, tandem or MS/MS spectrum a real CID spectrum While dependent upon the particular goal of your analysis, the MS is usually programmed to conduct a single MS scan followed by several MS/MS scans MS/MS scans are usually conducted on the x most abundant peptides (m/z), where x is 1-20 1 MS followed by 4-20 MS/MS scans (depending upon the instrument) is typical Step 3 The ion trap is emptied, refilled, and the process repeated - the entire MS-MS/MS cycle takes 1-4 seconds and is thus repeated thousands of times per MS analysis typical LC-MS run is 1-2 hrs average ~10,000 MS/MS per hour for a complex sample How does the MS/MS give you sequence information? the most common and informative fragment ions are generated by fragmentation of the amide bonds between amino acids b-ions if charge is retained by the amino-terminal fragment y-ions if charge is retained by carboxy-terminal fragment the differences in mass between the peptide fragments can be used to reconstruct the sequence of the original (parent) peptide (this is called de novo sequencing) but fragmentation pattern matching is used more often (we will talk about this later) a real CID spectrum getting your sequence – most of the time, we use database searching a user-defined protein database is subjected to in-silico digestion with the appropriate protease(s) to generate a list of all possible peptides a theoretical fragmentation pattern is then generated for each peptide parent ion mass (MS) and fragmentation data (MS/MS) from your analysis are compared to the theoretical data to find the best match matches may then be subjected to statistical analysis to determine the quality of the ID (p-value) spectral matching is also becoming more popular millions of spectra have been generated and searched already can keep these spectra in a library, then search for the best match to our newly generated spectra in the library advantages – can identify “messier” spectra, and is very fast disadvantges – if your peptide of interest has not been observed before, it won’t be in the library, and may not be compatible between different machine types Real spectral matching Mass spectrometry identification of proteins protein peptides Proteolytic digestion LC separation 12 14 Time (min) 16 Peptide selection; fragmentation Protein identification Peptide identification Database searching 200 400 600 800 10001200 m/z m/z putting it all together identification of peptides tells you which proteins were in your sample in the first place can identify hundreds of proteins in a single MS run can identify thousands of proteins in multiple MS runs of fractionated samples questions? take a break MBP 1001 Lecture Part 2 Okay, so I understand how to identify peptides - and therefore proteins - so what? i.e. what can proteomics do for you? some typical proteomics goals: global protein analysis protein machines protein-protein interactions PTMs quantitation global protein analysis goal - identification of every protein in a cell, tissue or organism - can compare state A to state B e.g. growth conditions, developmental stages, +/- hormone, mitogen or stress normal vs. disease state? typically involves extensive upstream protein (or peptide) fractionation however, some issues: dynamic range (MS vs serum?) massive amounts of machine, computer, and analysis time what proteins are present in each organelle? protein-protein interactions most cellular processes are carried out by multiprotein complexes (think transcription, translation, mRNA splicing, proteosomal degradation) to know your friends is to know you: interacting partners provide invaluable insight into understanding protein function and regulation interacting partners also change in response to signaling events, providing further clues to function signaling or metabolic pathways function in a stepwise fashion understanding how these pathways are structurally connected tagged protein/MS analysis - general tag protein of interest expression in relevant cell/tissue isolation optional sample fractionation MS identification epitope tagging short AA sequence recognized by Ab - FLAG, HA, GluGlu, etc. metal binding - 6xHis calcium binding - CaM other strong bimolecular interactions: biotin/avidin, GST/glutathione, chitinBP/chitin, MBP/maltose TAP (tandem affinity purification) consists of two protein tags, usually separated by a protease cleavage site *how might a tag affect protein-protein interactions? *pros/cons of different tag types? tandem affinity purification (TAP) strategy 1 express POI as a fusion with 2 peptide tags ProtA CaMBP protein of interest interacting partners 2 bind to IgG matrix, cleave with Tobacco Etch Virus protease TEV TAP tag strategy (step 2) 3 bind to calmodulin matrix CaM 4 elute EDTA CaM 5 identify co-purifying proteins large-scale tagging projects several large-scale tagging/MS projects now published have identified thousands of novel protein-protein interactions good: pull down multiprotein complexes, providing a more realistic picture of interactions possible to see interactions that are dependent upon PTMs can do this type of analysis in relevant organism/cell/tissue not so good: lots of non-specific interactions; with sepharose, tags, or due to overexpression detection of low abundance proteins may require scale-up *how might you deal with these problems? other problems with large-scale techniques? all of these techniques are biased toward proteins of higher abundance -many low stoichiometry interactions may be missed -usually conducted under a single condition, may miss very interesting regulated interactions large-scale take-home messages large-scale prot-prot interaction techniques are extremely valuable for obtaining a snapshot in time, and under a given set of environmental/ developmental conditions this knowledge is extremely valuable - connects formerly unconnected pathways and processes provides an overview of how protein machines are built and interact with each other however -not much fine detail in these studies, much of the data uncorroborated by other methods -if you are interested in a particular protein, protein machine, or biochemical pathway, present large-scale data will likely be unsatisfactory -for these types of questions, more focused studies are required directed studies IPs and tagged proteins high density prot-prot interaction networks small-scale quantitative proteomics classical IP analysis of protein complexes experimental control samples are cleaned up until maximal difference between sample and control is achieved: *pros/cons? 116 kD 97 kD 66 kD 45 kD weak interactors are lost lots of background extensive optimization required conditions vary for each sample specificity of Ab? what kind of control(s)? what does my protein do? generating a high-density interaction map you have found an interesting protein of unknown function what does it do? protein phosphatase 2A (PP2A or PPP2) major Ser/Thr phosphatase in mammalian cells conserved from yeast to human PPP2 functions in most cases as a trimeric complex numerous regulatory subunits (B) thought to confer substrate specificity catalytic (C) regulatory (B) adapter (A) additional human PP2A-related phosphatases two additional phosphatases highly related to PPP2C PPP4C is 67% identical to PPP2C PPP6C is 58% identical to PPP2C molecular organization of PPP4 and PPP6 was unknown who do PPP4 and PPP6 talk to? PPP2 PPP2 catalytic (C) regulatory (B) PPP2 adapter (A) PPP4 catalytic (C) ? PPP6 catalytic (C) ? Generating a human protein interaction network Clone protein of interest into a TAP-tag vector A Stably express TAP-tagged proteins in human 293 cells B C F E G H D Harvest cells, and affinity-purify recombinant proteins, as well as associated proteins Identify all proteins in the complex by mass spectrometry Obtain the cDNA for each protein identified I high density data via iterative TAP-tagging reveals mutually exclusive and cooperative interactions in the PPP2 module 2 PPP2R2 1 IGBP1 PPP2C PPP2R1 3 PPP2R5 PTMs PTMs commonly identified using MS phosphorylation ubiquitylation glycosylation methylation acetylation hundreds of others… identified primarily via a mass shift of a particular amino acid Reading a CID spectrum i. unmodified peptide ii. phosphopeptide iii. sumoylated peptide enrichment of phosphopeptides IMAC immunocapture chemical capture affinity chromatography identification of a Ub conjugation site quantitation and mass spectrometry two primary methods spectral counting - characterizing the number of spectra observed for a given protein, in relation to other proteins, or between samples stable isotopes (e.g. 13C, 15N) incorporation of stable isotopes into peptides does not alter biochemical properties (e.g. chromatography is unaffected) but changes the mass of the peptide - this, of course, is a property that the MS can see quantitative proteomics with stable isotopes intensity Isotopic mass difference intensity is proportional to peptide abundance m/z Isotope-coding Separation Quantitation Identification MS “heavy” peptide “light” peptide LC intensity intensity MS/MS m/z m/z spectral counting in a series of AP-MS analyses protein A was tagged and isolated, sample subjected to LC-MS/MS data protein A B C D condition 1 condition 2 protein B knockout 684 599 620 131 157 0 176 10 204 34 0 0 what can you get from this data? isotopic labeling strategies Metabolic labeling: SILAC Cells grown in “light” SILAC Fractionation LC-MS/MS lysis Affinity purification Cells grown in “heavy” SILAC Labeling with “light” ICAT Lysis Proteolytic digestion Chemical labeling: ICAT Fractionation LC-MS/MS Affinity purification Labeling with “heavy” ICAT Proteolytic Isolation of digestion ICAT-labeled peptides absolute quantitation what if you would like to know absolute levels of your protein/peptide? e.g. determine stoichiometries of various proteins in protein complexes? AQUA – peptides synthesized with stable isotopes, to use as internal standards spiked into sample, and used to quantify endogenous peptide by comparing ion intensities can be made with standard PTMs END iTRAQ 0 min 60 min 30 min 120 min treat cells A C C A A B A isolate complex B proteolyze iTRAQ label iTRAQ 114 iTRAQ 115 iTRAQ 116 iTRAQ 117 combine quantitate and identify