Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Degradomics wikipedia , lookup
Structural alignment wikipedia , lookup
Protein domain wikipedia , lookup
Circular dichroism wikipedia , lookup
Intrinsically disordered proteins wikipedia , lookup
Protein folding wikipedia , lookup
Bimolecular fluorescence complementation wikipedia , lookup
Protein design wikipedia , lookup
Protein purification wikipedia , lookup
Protein–protein interaction wikipedia , lookup
Western blot wikipedia , lookup
Protein structure prediction wikipedia , lookup
Nuclear magnetic resonance spectroscopy of proteins wikipedia , lookup
Homology modeling wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
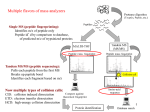
Facts and Fallacies about de Novo Sequencing & Database Search 1. There are a large number of high quality spectra left unassigned after DB search. True False Unassigned Spectra in ABRF/iPRG 2011 Study Unassigned Spectra PEAKS DB De novo sequencing PEAKS PTM SPIDER • • • • Nonspecific trypsin cleavages Novel peptide/incomplete database PTM Mutations 2. Nonspecific cleavage, PTM, mutations and novel peptides are the main reasons for the unassigned spectra. True False Average Software Misses Peptides 3. De novo sequencing is slow. True False Speed • PEAKS 6 de novo sequence 15 spec/second. – Intel i7 Quad Core, 8GB RAM. – Trypsin – Orbitrap CID MS/MS, mostly charge +2/+3 • PEAKS 7 (coming soon): – Improve speed on high charge states and longer peptides. – Add 8 core support in standard (desktop) license. 4. De novo should be done after DB search. True False DB search DB peptides Unassigned spectra de novo seq. de novo peptides Order of de Novo and DB • Better conduct de novo on all spectra. – De novo not slow, and computing is cheap. – De novo provides independent validation for DB result. # consensus AA (de novo vs. DB search) false without with de novo de novo true true score 5. My protein sequence is confirmed with two unique peptide hits. True False Routine Full Protein Coverage • For regular proteins, full sequence coverage can be routinely achieved with – 3 or more enzyme digests, and – multiple algorithms in PEAKS 6. • For highly variable proteins (such as antibodies), BSI offers data analysis service for antibody sequencing. 6. If a peptide is identified with 1% FDR, then it’s sequence is 99% correct. True False Peptide Validation vs. Amino Acid Validation You are confident about the peptide sequence only if • you can de novo sequence it, and • the de novo sequence matches the database peptide. 7. I don’t need de novo sequencing if I have a protein DB. True False 8. Target-decoy provides a reliable result validation for every DB search engine. True False Target-Decoy Incompatible with Certain Highly Optimized Search Engines weak hits confident protein weak protein • Adding “protein bonus” to peptide hits increases accuracy. • But it creates bias between target and decoy. – In extreme, bonus is so large that only peptides from target proteins are selected. – This gives the wrong impression that FDR=0, while there are still false peptides in the result. Decoy Fusion Is A More Powerful Validation Method weak hits confident protein weak protein • Decoy fusion append a decoy sequence to each protein. • Recreates the balance. • The built-in validation method since PEAKS 5.3. 9. Combining 1% FDR results of multiple engines gives 1% FDR. True False Error Accumulation Target(decoy) FDR% PEAKS DB 3870(38) 1% Mascot 2369(23) 1% Correct < sum of the two Error ≈ sum of the two PEAKS DB 1696(37) 2.4% Mascot 2174(1) 0.1% 195(22) 13% Combined FDR = 1.5% • In PEAKS, the inChorus algorithm automatically selects a less than 1% common FDR for each engine so that the combined FDR is approximately 1%. 10. There is no automated way to validate de novo sequencing results. True False