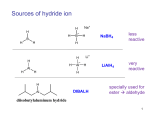
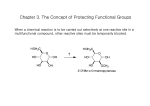
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Enantioselective synthesis wikipedia , lookup
Kinetic resolution wikipedia , lookup
Marcus theory wikipedia , lookup
Hydroformylation wikipedia , lookup
Physical organic chemistry wikipedia , lookup
Ring-closing metathesis wikipedia , lookup
Wolff–Kishner reduction wikipedia , lookup
Hofmann–Löffler reaction wikipedia , lookup
Stille reaction wikipedia , lookup
Tiffeneau–Demjanov rearrangement wikipedia , lookup
Bottromycin wikipedia , lookup
Elias James Corey wikipedia , lookup
Vinylcyclopropane rearrangement wikipedia , lookup
Discodermolide wikipedia , lookup
Nucleophilic acyl substitution wikipedia , lookup
Wolff rearrangement wikipedia , lookup
Petasis reaction wikipedia , lookup
1348_C69.fm Page 1 Monday, October 13, 2003 3:22 PM 69 Photoremovable Protecting Groups 69.1 Introduction ..................................................................... 69-1 69.2 Historical Review.............................................................. 69-2 o-Nitrobenzyl • Benzoin • Phenacyl • Coumaryl and Arylmethyl 69.3 Carboxylic Acids............................................................. 69-17 o-Nitrobenzyl • Coumaryl • Phenacyl • Benzoin • Other Richard S. Givens University of Kansas Peter G. Conrad, II University of Kansas Abraham L. Yousef University of Kansas Jong-Ill Lee University of Kansas 69.4 Phosphates and Phosphites ........................................... 69-23 o-Nitrobenzyl • Coumaryl • Phenacyl • Benzoin 69.5 Sulfates and Other Acids................................................ 69-26 69.6 Alcohols, Thiols, and N-Oxides .................................... 69-27 o-Nitrobenzyl • Thiopixyl and Coumaryl • Benzoin • Other 69.7 Phenols and Other Weak Acids..................................... 69-36 o-Nitrobenzyl • Benzoin 69.8 Amines ............................................................................ 69-37 o-Nitrobenzyl • Benzoin Derivatives • Arylsulfonamides 69.9 Conclusion...................................................................... 69-40 69.1 Introduction Photoremovable protecting groups are enjoying a resurgence of interest since their introduction by Kaplan1a and Engels1b in the late 1970s. A review of published work since 19932 is timely and will provide information about several new groups that have been recently developed. The scope of this review is, therefore, limited to recent developments in the field and will cover only the applications with major functional groups that have been “protected” by a photoremovable chromophore. The review is not intended to be comprehensive but focuses instead on a series of well-chosen examples of chromophores that were deployed as protecting groups with a select group of representative functional groups. Because the focus of this review is the application of photoremovable protecting groups, emphasis is placed on synthesis of the protected functionality and on the procedures employed for deprotection, including the protection and photodeprotection yields, the deprotection reaction rates, and the quantum efficiencies, when available. An attempt has been made to list the advantages and disadvantages of each photoremovable protecting group as well as a brief discussion of the mechanism for the photodeprotection. 0-8493-1348-1/04/$0.00+$1.50 © 2004 by CRC Press LLC 69-1 1348_C69.fm Page 2 Monday, October 13, 2003 3:22 PM 69-2 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition When the literature is insufficient for providing a comprehensive treatment of applications of a photoprotecting group, then only a brief discussion is provided. An exhaustive list of applications for any of the chromophores is not included; these may be found by consulting other reviews or the original literature on a topic. Several good reviews on photoremovable protecting groups have appeared since this topic was reviewed in 1993 (e.g., Adams and Tsien3 and Corrie and Trentham4). Notable among the more recent reviews are those by Wirz,5 Bochet,6 and Givens.7 A volume of Methods in Enzymology devoted entirely to the chemistry and applications of photoremovable protecting groups, also termed “caged” compounds, that are employed in biochemistry and other biological studies has also appeared. In general, photolysis reactions present a noteworthy and often ideal alternative to all other methods for introducing reagents or substrates into reactions or biological media. The ability to control the spatial, temporal, and concentration variables by using light to photochemically release a substrate provides the researcher with the ability to design more precisely the experimental applications in synthesis, physiology, and molecular biology. Among the many possible examples is the recently reported inhibition–reactivation of protein kinase A by photolysis of the dormant enzyme.8–10 In this demonstration, it is necessary that the deprotection process be initiated by photolysis of the dominant chromophore of the protecting group. Covalent blocking of the functional groups at the active site of an enzyme essentially suspends its mode of action and virtually shuts down the catalytic cycle. It is this feature that has attracted biochemists to the use of protecting groups for the investigation of biological mechanisms. In synthesis, the protecting group serves as a mask that renders a functional group inert to subsequent synthetic reaction conditions,11 except, of course, conditions that are required for the removal of the protecting group. Construction of combinatorial platforms with photoremovable linkers is just one example of the applications in synthesis. Photorelease is sometimes termed a traceless reagent process because no reagents other than light are needed. The advantage of a process that requires no further separation of spent reagents is attractive. There are several limitations to the use of commonly employed protecting groups in synthesis and for mechanistic studies of biological processes. The reactions for incorporating and subsequently removing protecting groups often involve acid or base that may be too harsh and interfere with the normal processes or otherwise be incompatible with the chemistry or biology under investigation. In mechanistic biochemistry, it is often the case that the typical hydrolysis deprotection reaction is far too slow to serve as a means of investigating the initial rates of reaction for rapid biochemical processes. An ideal remedy to these limitations is a protecting group that could be removed under neutral buffered aqueous conditions, thus avoiding any alterations to the substrate or to the natural biological environment.12 The release should occur on a time scale fast enough for kinetic analysis of any subsequent rapid biological processes. Such a group may be a photoremovable protecting group. 69.2 Historical Review In 1962, Barltrop et al.13 were among the first to report a photochemical deprotection reaction of a biologically significant substrate; here, glycine was released from N-benzyloxycarbonyl glycine: O O N H OH O hν CH3 + OH H2N O + CO2 (69.1) This seminal discovery prompted the development of several additional photoremovable protecting groups. The success of many researchers in biology, particularly Kaplan,1a led to the description of the photoactivatable group as a “cage” to describe its deactivating influence on the biological substrate to which it is covalently attached.14–17 Ideally, the cage detaches only through the action of light. 1348_C69.fm Page 3 Monday, October 13, 2003 3:22 PM 69-3 Photoremovable Protecting Groups It is important that the photoremovable protecting group also possess several other desirable properties. The properties were originally compiled by several researchers in the field, including Sheehan and Umezawa12 and Lester and Nerbonne,18 who provide a series of benchmarks for evaluating the efficacy of a photoremovable group in a given circumstance or for evaluating the potential of a new cage chromophore. A more useful adaptation of the Lester rules and Sheehan criteria includes the following: 1. The substrate, caged substrate, and photoproducts have good aqueous solubility for biological studies. For synthetic applications, this requirement is relaxed. 2. The photochemical release must be efficient (e.g., Φ > 0.10). 3. The departure of the substrate from the protecting group should be a primary photochemical process (i.e., occurring directly from the excited state of the cage chromophore). 4. All photoproducts should be stable to the photolysis environment. 5. Excitation wavelengths should be longer than 300 nm and must not be absorbed by the media, photoproducts, or substrate. 6. The chromophore should have a reasonable absorptivity (a) to capture the incident light efficiently. 7. The caged compounds, as well as the photoproduct from the cage portion, should be inert or at least benign with respect to the media, other reagents, and products. 8. A general, high-yielding synthetic procedure for attachment of the cage to the substrate must be available. 9. In the synthesis of a caged substrate, the separation of caged and uncaged derivatives must be quantitative. This is also necessary for the deprotection process for synthetic applications. While these are the desirable guidelines for an ideal photoremovable protecting group, a potential cage that lacks one or two of these properties may still be very useful; however, the absence of several of these features may militate against the use of that group as a photoremovable protecting group for a specific application. Some representative examples of photoremovable protecting groups that qualify as meeting the Lester and Sheehan criteria include α-substituted acetophenones, benzoins, benzyl groups, cinnamate esters, coumaryl groups, and, the most popular of them all, the o-nitrobenzyl esters and their analogs. o-Nitrobenzyl It was also Barltrop et al.19 who first reported the use of an o-nitrobenzyl group to release benzoic acid (see Eq. (69.2)). The poor yield stemmed from the subsequent conversion of 2-nitrosobenzaldehyde (3), the initial photoproduct, into azobenzene-2,2′-dicarboxylic acid (4),20 which then competed for the incident light. Yields were dramatically improved with the use of α-substituted nitrobenzyl esters (75 to 95% conversion), as seen from 5 in Eq. (69.3). The resulting photoproduct from o-nitrobenzyl ester 5 was a less reactive nitroso benzophenone derivative. O O O hν HO2C O OH + N H NO2 NO 3 2, 17% 1 N CO2H 4 (69.2) Ph O O hν O 2 Ph + NO2 5 NO 6 (69.3) 1348_C69.fm Page 4 Monday, October 13, 2003 3:22 PM 69-4 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition The report of the release of ATP by this method appeared in a 1978 rate study reported by Kaplan and co-workers.1a Inorganic phosphate (Pi) and ATP were released from their 1-(2-nitrophenyl)ethyl (NPE) and 2-nitrobenzyl (NB) esters, respectively: R O OP R' O NO2 7a 7b 7c 7d O- O hn, 342 nm R = H, R' = R = H, R' = O-ADP R = CH3, R' = OR = CH3, R' = O-ADP R O OP R' O + NO 3 R = H, CH3 P R' = OATP R' = O-ADP i (69.4) The results of the release of Pi from NPE and NB showed very similar quantum efficiencies of 0.58 and 0.50, respectively; however, the release of ATP from the two cages gave very different rates of conversion. NPE released 80% of the caged ATP in less than 60 s compared with 25% for release from the NB caged ATP. These results further indicated that the α-substituted nitrobenzyl esters were better suited as phototriggers. Kaplan’s investigation also explored the potential use of photoprotecting groups in a physiological environment. Na+,K+-ATPase, the enzyme responsible for sodium/potassium transport through cell walls, served as the model for exploring the effect of the caged ATP (NPE-ATP) on the Na+:K+ transport associated with enzymatic activity. The enzyme acquires ATP as the energy source through hydrolysis of the terminal γ-phosphate. The hydrolytic activity of the enzyme can be monitored by the detection of Pi generated from the free ATP consumed by the enzyme. In the absence of photolysis, NPE-ATP was shown to be resistant to hydrolysis by the enzyme. Upon photolysis, the liberated ATP triggered the response of the enzyme and Pi release was observed. The successful introduction of o-nitrobenzyl caged ATP into physiological media instigated interest in expanding the applications of caged release to a wide variety of biochemical systems. The list includes the mechanism of release of Pi in skeletal muscle,21 the function of cAMP in the relaxation of distal muscle,22 the ATP-induced mechanism of actomyosin in muscle contraction,23 and the activation of antitumor antibiotics to highly reactive pyrrolic-type intermediates responsible for DNA crosslinking reactions.24 Benzoin Sheehan and Wilson25 were the first to explore the photochemical rearrangement of certain benzoin derivatives to yield 2-phenylbenzofuran (9). These rearrangements occurred with concurrent loss of groups attached α to the carbonyl just as in the case of the α-chloroacetophenones. They suggested that benzoins, especially the 3′,5′-dimethoxybenzoin chromophore, could serve as a photoremovable protecting group for carboxylic acids. In 1984, Givens et al.26 showed that phosphates were quantitatively expelled from the ungarnished benzoin cage, as shown in Eq. (69.5), thus extending the range of applications and the nature of the parent chromophore. The only major product accompanying the released phosphate 1348_C69.fm Page 5 Monday, October 13, 2003 3:22 PM 69-5 Photoremovable Protecting Groups TABLE 69.1 Quantum Efficiencies for Benzoin Phosphate Esters 8a–c Phosphate Ester 8a 8b 8b 8c 8c Solvent pH Φdis Φfuran Φphosphate C 6H 6 H2O/CH3CN H2O/CH3CN H2O/CH3CN H2O/CH3CN nd 2.0 7.0 2.0 7.0 0.28 0.37 nd 0.38 nd 0.26 0.20 0.07 0.14 0.08 nd 0.12 0.013 0.15 0.01 Note: All reactions were run in 60% aqueous acetonitrile, except 8a, as indicated; phosphate esters were irradiated at 350 nm and monitored via 31P NMR; nd = not determined. Source: Adapted from Givens et al.27 O hn O 3 H ST PO(OR)(OR') 3 8 <20 ns 3 O O + H O PO(OR)(OR') 8a R = R' = Et 8b R = iPr, R' = Na+ or H + 8c R = R' = +Na or +H 310, 670 ns ST <5 ns 1 10 CF3CH2OH O Ph 9 O OCH2CF3 + O P OR' O OR from laser flash studies R = R' = Et R = iPr, R' = +Na or +H R = R' = +Na or +H, (Pi) (69.5) was 2-phenylbenzofuran 9. These reactions were quenched with naphthalene, piperylene, or, for aqueous studies, sodium 2-naphthalenesulfonate, all well-known triplet quenchers; this established the short-lived triplet (3 to 14 ns) as the reactive excited state for benzoin. Further information was revealed from Stern–Volmer quenching analyses which provided the rate of release of phosphate from the benzoincaged ester. Extremely fast rates (kr > 108 s–1) were measured along with good efficiencies for the reaction, ranging from 28 to 38% (Table 69.1).27,28 Phosphorescence spectra supported the multiplicity assignments and also established the triplet energy at 73 ± 1 kcal/mol. The efficiencies for the disappearance of the caged phosphates (Φdis), as well as the appearance of 2phenylbenzofuran (Φfuran) and phosphate (Φphosphate), were determined to be pH dependent with higher efficiencies reported under acidic conditions. The greater efficiency at lower pH suggests that the protonated phosphate is a more favorable leaving group than its conjugate base. This study was extended to nucleotide release from the benzoin cage through the synthesis and photolysis of benzoin cAMP. The efficient release of cAMP as the exclusive product with quantum efficiencies on the same order of magnitude as the model phosphate esters first demonstrated the application of benzoin as a cage for 1348_C69.fm Page 6 Monday, October 13, 2003 3:22 PM 69-6 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition TABLE 69.2 Quantum Efficiencies for Photolysis of Benzoin Adenosine Cyclic 3′,5′-Monophosphatea Aqueous Buffer pH Φdis Φfuran ΦcAMP Tris (D2O) Tris (H2O) Phosphate (D2O) Phosphate (H2O) Perchloric (D2O) 7.3 7.3 8.4 8.4 1.6 0.39 0.37 nd nd 0.40 0.19 0.17 0.17 0.17 0.16 0.34 0.34 nd nd 0.36 a Irradiations were carried out in 1:1 buffer: 1,4-dioxane at 350 nm. Quantum efficiencies (Φ) were measured using 31P NMR, except where indicated (nd). Source: Adapted from Givens et al.28 * OMe OMe O O hν OMe O OMe O R R O O * MeO H MeO H OMe O demotion H O H R O O R O H OMe OMe O O O OMe O + O R OMe OMe 11 SCHEME 1 Benzoin photorelease mechanism. nucleotides (Table 69.2). 31P spectra of released cAMP demonstrated that cAMP was the only phosphate present after release. As Table 69.2 indicates, the quantum efficiencies remained relatively constant throughout the pH range examined. It has been determined that carboxylate derivatives are released more readily when there are electrondonating substituents at the meta positions of the benzyl ring.29 The absorption spectra of the benzoin esters along with the observations that cyclization was enhanced by meta electron donating groups led observers to believe that reaction was taking place through an n,π* singlet state (see Scheme 1). It was suggested that the excitation of the phenacyl group led to a short-lived 1,3-biradical, followed by demotion to the ground state. The zwitterionic intermediate led directly to the loss of the leaving group. Aromatization through loss of a proton gave the benzofuran 11 as the principal photoproduct. The inability to quench the reactions with high concentrations of piperylene suggested that the reaction originated from the singlet excited state or, alternatively, from a very short-lived, unquenchable triplet. 1348_C69.fm Page 7 Monday, October 13, 2003 3:22 PM 69-7 Photoremovable Protecting Groups Based on these observations Corrie and Trentham4 re-examined the photochemistry of several substituted benzoin phosphates. They found that the 3′,5′-dimethoxybenzoin cage was best suited among those investigated for the release of Pi. The formation of 3′,5′-dimethoxy-2-phenylbenzofuran (11) occurred at a rate that exceeded 105 s–1 and a quantum efficiency of 0.78. While the rate of product formation was lower than that reported by Givens and Matuszewski,26 the efficiency for the substituted benzofuran analog was much higher. For the benzoin series, the primary photoproduct 11 is also a strongly absorbing chromophore and thus competes for incident light and forms photodimers along with several other unidentified products upon further irradiation. Yet another limitation of this system is the presence of a chiral center alpha to the carbonyl, which engenders isolation and purification problems in the synthesis of benzoin-protected chiral substrates. More recently, Rajesh et al.30 examined the earliest events in the photolysis of benzoin diethyl phosphate (8a). With nanosecond resolution, the LFP excitation of 8a in trifluoroethanol gave an immediate, permanent 300-nm absorption identified as the benzofuran photoproduct (Eq. (69.5)). A second transient absorption at 570 nm was also observed which decayed with a first-order rate constant of ~2 × 106 s–1 in degassed acetonitrile or trifluoroethanol that was assigned to the triplet α-ketocation 10. The intermediate could be trapped by trifluoroethanol, yielding trifluoroethyl benzoin ether. Evidence for the intermediacy of the α-ketocation triplet came from experiments with added halide ion or azide in which electron transfer quenching of the transient was observed. Oxygen and naphthalene quenching experiments demonstrated that 310 was formed adiabatically on the triplet manifold from 38a. Temperature-dependent studies indicated an activation energy for decay of 310 of 8.6 kcal/mol to the singlet, which then reacted with trifluoroethanol to form the trifluoroethyl ether. Stern–Volmer analysis of the naphthalene quenching gave a triplet lifetime for 38a of τ3 = 18 ns. The formation of 2-phenylbenzofuran during the nanosecond laser flash experiment was corroborated by a picosecond study of 8a. A rise time of 2 to 4 ps was determined for the 340 nm transient. A rich fluorescence emission obtained in the nanosecond study was shown to arise from 19 generated during the nanosecond laser excitation pulse. Naphthalene also quenched the formation of 9 at the same rate as the formation of 310, establishing that the two primary photoproducts came from the same triplet (i.e., 38a). Thus, for the unsubstituted benzoin phosphates, reaction proceeds exclusively through the triplet manifold. Phenacyl In a similar study, Sheehan and Umezawa12 employed a stripped-down version of the benzoin chromophore, the p-methoxyphenacyl group, for the release of benzoic acid, several amino acid derivatives, and peptides, as shown in Eq. (69.6) and Table 69.3. O O hν, pyrex R ethanol or dioxane MeO 12 + free acid MeO 13 (69.6) In this case, the photoproduct was the p-methoxyacetophenone (13), a reduction product. The proposed mechanism (Scheme 2) was a simple homolysis of the carbon-oxygen bond. Ethanol serves as a hydrogen atom donor during this process, and in the presence of 1 M benzophenone or naphthalene the reaction was completely quenched, indicating a triplet reaction pathway. Benzophenone and naphthalene are known quenchers of acetophenones and have triplet energies of 68 and 62 kcal/mol, respectively. Epstein and Garrossian31 reported the release of ethyl and phenyl phosphate esters from the corresponding p-methoxyphenacyl phosphates in 1,4-dioxane. The released phosphates were recovered in high yields (Et, 86%; Ph, 74%) along with 13 (91–84%) as the only observed photoproducts. The absence of any rearranged ester products contrasted with reports by Anderson and Reese34 for substituted αchloroacetophenones (vide infra). The rationale for the discrepancy advanced by Epstein was an altered 1348_C69.fm Page 8 Monday, October 13, 2003 3:22 PM 69-8 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition TABLE 69.3 Percent Yield for the Release of Various Acidsa from the Corresponding 4-Methoxyphenacyl Esters (12) R Solvent Irradiation Time (hr) Yield of ROOH (%) PhCOO PhCOO Boc-L-Ala Boc-L-Ala Boc-Gly Boc-L-Phe Z-D, L-Ala Phthaloyl-Gly Tri-Gly Z-L-Trp Z-Gly-Gly Z-L-Asp(OBz)-L-Ser Dioxane Ethanol Dioxane Ethanol Ethanol Dioxane Dioxane Dioxane Dioxane Ethanol Ethanol Dioxane 17 6 17 6 6 17 6 17 17 4 5.5 9 81 96 82 93 94 89 84 80 58 33 77 49 a All reactions were carried out below room temperature at (5 × 10–3–10–2) M using a Pyrex filter. Irradiations were complete in 6 hr in ethanol and 11–17 hr in dioxane. Yields were determined from product isolation following photolysis. Source: Adapted from Sheehan and Umezawa.12 SCHEME 2 Sheehan et al. mechanism mechanism due to a change in solvent. The solvent 1,4-dioxane may not be sufficiently polar to support the formation of the zwitterionic precursor to the Favorskii-like rearrangement that was proposed by Anderson and Reese. This was required for the rearrangement to the spirodienedione intermediate. Reinvestigation of the photolytic cleavage using polar solvents would have been an interesting test of this hypothesis. (69.7) Givens et al.28 examined the photorelease of phosphate esters using t-butyl alcohol and methanol as solvents, the former being a poor hydrogen atom donor and both exhibiting increased polarity compared with 1,4-dioxane. The results correlated well with those of Anderson and Reese in that the major photoproduct was the rearranged ester, not the photoreduction product (Eq. (69.7)). The amount of 13 was decreased to 21% in methanol and 14% in t-butyl alcohol. Further investigation of the solvent dependency for the release of phosphates revealed a solvent isotope effect with deuterated vs. protiated methanol as the solvent (Table 69.4). The formation of p-methoxyacetophenone was suppressed by a factor of five when 1348_C69.fm Page 9 Monday, October 13, 2003 3:22 PM 69-9 Photoremovable Protecting Groups TABLE 69.4 Quantum Efficiencies and Solvent Isotope Effects (kH/kD) for Photolysis of 4-Methoxyphenacyl Diethyl Phosphate (14a)a Φ14a Φ15 kH/kD15 Φ13 kH/kD13 0.036 0.42 — — 0.026 0.20 0.14 0.11 — — 1.4 1.8 0.074 0.07 0.013 0.053 — — 5.4 1.3 Solvent C6H6, t-BuOH (3:1) CH3OH CD3OD CH3OD a Irradiations were performed in the indicated solvent at 300 nm. kH/kD is a relative efficiency for H vs. D abstraction. Error limits are ±10%. Source: Adapted from Givens et al.28 TABLE 69.5 Photolysis of α-Chloroacetophenones: Yields Obtained for Photoproducts 17 to 20 Under Varying Conditionsa Methanol Acetone(aq) Acetonitrile(aq) 16 Ar 17 19 17 18 20 17 18 20 a b c d Phenyl 4-Methyl phenyl 4-Methoxy phenyl 4-Chloro phenyl 60 62 35 60 00 06 34 00 37 24 16 10 24 47 43 14 29 23 14 31 14 14 09 06 47 70 54 35 18 11 08 18 a Photolysis of 2% degassed solution of 16a–d in methanol, 95:5 acetone/ water, or 95:5 acetonitrile/water using 300-nm lamps was carried out in the presence of propylene oxide as a halogen scavenger. Irradiations were 4 hr and yields are isolated products. Source: Adapted from Dhavale et al.32 photolysis was carried out in CD3OD compared with either CH3OD or CH3OH, suggesting that a ratedetermining hydrogen abstraction occurs in the photoreduction process. Sheehan first suggested this mechanistic pathway (vide supra). Indeed, Dhavale et al.32 have shown that the ratio of rearrangement to reduction for substituted αchloroacetophenones is solvent dependent [Eq. (69.8) and Table 69.5]. Dhavale’s group reported that irradiation of substituted α-choroacetophenones in methanol resulted in more photoreduction, whereas in aqueous acetonitrile rearrangement to the phenylacetate esters became the major pathway. For a given solvent, the ratio of rearrangement to reduction increased with the electron-donating power of the substituent. hν O Cl Ar 16 solvent O Ar CH3 17 + O OR Ar + O 18 R = H 19 R = CH3 Ar Ar O 20 (69.8) Scheme 3 outlines Dhavale’s proposed mechanism for the chloride loss and subsequent rearrangement, beginning with carbon–chlorine bond homolysis. An electron transfer from the α-ketoradical to the chlorine atom leads to the ion pair 21. The ion pair is more susceptible to Favorskii-like rearrangement in polar solvents; therefore, the rearranged phenylacetic acid is favored in polar, protic solvents. Hydrogen abstraction, resulting in the formation of 17, prevails in those solvents that are good hydrogen atom donors. Sonawane et al.33 investigated the photorearrangement of several para-substituted propiophenones as a convenient entry for substituted α-arylpropionic acids, as shown in Eq. (69.9): 1348_C69.fm Page 10 Monday, October 13, 2003 3:22 PM 69-10 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition O O O Cl hν H-abstraction X CH3 Cl X + HCl X 16 17 hν single electron transfer (SET) O H X H OH a Favorskii-like O Cl 1,2-aryl migration Cl O X X 21 18 SCHEME 3 The mechanism suggested by Dhavale et al.32 TABLE 69.6 Product Distribution from Photolysis of 22a–i in Aqueous Acetone or Aqueous Methanola Substrate Acetone(aq) Methanol(aq) 22 X 23 24 25 23 24 26 a b c d e f g h i H CH3 C 2H 5 n-C3H7 i-C4H9 t-C4H9 Cl Ph OCH3 58 84 82 84 74 78 45 40 32 25 5 6 5 10 7 25 25 10 — — — — — — 20 35 50 39 76 74 77 65 69 30 18 80 30 8 9 9 15 8 51 26 12 — — — — — — 30 35 70 a Irradiations were carried out in 95:5 solvent/water solutions employing a Hanovia 200-W, mediumpressure mercury vapor lamp with a Pyrex filter until the complete disappearance of starting material. The photoreaction was monitored and yields were determined with GLC and 1H NMR. Source: Adapted from Sonawane et al.33 O Cl X 22a-i solvent propylene oxide O O hν, 300 nm OR OH O X 23 + + X X 24 25 R = H 26 R = CH3 (69.9) In almost every case, para-substitution promoted rearrangement (Table 69.6). Phenyl- and chloro-substitutions in the para position were the only cited examples where rearrangement did not dominate in methanol. These cases also showed significant reduced and hydrolyzed products. 1348_C69.fm Page 11 Monday, October 13, 2003 3:22 PM 69-11 Photoremovable Protecting Groups TABLE 69.7 Product Formation from Photolysis of Substituted Phenacyl Chloridesa Aryl Substitution, X % Ethyl Aryl Acetate % Acetophenone 32 32 — 32 4 — — — — — 26 30 3 16 58 53 48 55 45 15 p-OH (27) p-OMe (16c) o-OH o-OMe p-Me (16b) H p-CO2Me p-Cl (16d) o-Cl m-OMe a Photolyses were carried out in 1% alcoholic solutions using a 500-W Hanovia mercury arc lamp. Reactions were carried out for 1 to 2 hr. Products were isolated via vapor phase chromatography. Source: Adapted from Anderson and Reese.34 While not purported to be a photoremovable protecting group, the study by Anderson and Reese34 on substituted phenacyl chlorides did reveal an interesting photochemical rearrangement for certain members of the series, particularly the report that the Favorskii-like rearrangement of p-hydroxyphenacyl chloride (27) in 1% aqueous ethanol gave two major photoproducts: p-hydroxyacetophenone (28) and ethyl p-hydroxyphenyl acetate (29), as shown in Eq. (69.10). The authors proposed a spiro intermediate 30. O O hν Cl OEt + 1% aqueous EtOH HO HO 28 27 O HO 29 O O 30 (69.10) Further examination of the proposed aryl participation hypothesis led to the observation that electrondonating groups in the ortho or para positions were necessary for the rearrangement to occur (Table 69.7). The results of Anderson and Reese coupled with the efficacy of the benzoin chromophore for cleavage of α substituents attracted our interest in developing the p-hydroxyphenacyl group as a photoremovable protecting group. We further rationalized that the introduction of a phenolic hydroxy group would enhance the aqueous solubility. The absence of the attendant α phenyl substituents on benzoin alleviated the stereogenic center problem present with benzoin derivatives. Furthermore, Anderson and Reese reported a Favorskii-like rearrangement of the chromophore, e.g., p-hydroxyphenacyl to p-hydroxyphenylacetate for 27 → 29 and for 16b,c, suggesting a significant hypsochromic shift in the chromophore. Thus the promise of p-hydroxyphenacyl as a possible phototrigger was too enticing to pass up. In 1995, we began a comprehensive exploration of a variety of p-substituted phenacyl phosphates for their efficacy toward releasing phosphate.2,28 Among the substituents examined, the p-acetamido, methyl p-carbamoyl, and n-butyl p-carbamoyl groups proved untenable because they gave a plethora of products, most of which resulted from coupling or reduction of an intermediate phenacyl radical [Eq. (69.11)]35,36 1348_C69.fm Page 12 Monday, October 13, 2003 3:22 PM 69-12 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition TABLE 69.8 Disappearance and Product Efficiencies for Ammonium Salts of p-Substituted Phenacyl Phosphate in pH 7.2 Tris Buffer at 300 nm p-Substituent 31a NH2 31b CH3CONH 31c CH3OCONH 14c CH3Oa 33 HOb 35 HOc a b c Φdis Φ34 <0.05 0.38 0.34 0.42 0.38 0.37 0.0 0.0 0.0 0.20 0.12 0.31 Φ32,13 Φother <0.05 0.11 Not determined 0.07 0.0 0.0 Not available Dimers Two unknowns Not available 0.0 0.0 Solvent was MeOH and diethyl phosphate was the leaving group. The diammonium salt of the mono ester; 10% CH3CN was added to the solvent. The ATP derivative. O O O X O P O hn, ROH O HO + X 31a-c, 14c O P O + other products O 32a-c, 13 a) X = NH2, b) X = NHCOCH3, c) X = NHCO2CH3, 14c) X = OCH3 (69.11) Table 69.8 gives the disappearance efficiencies for several p-substituted phenacyl phosphates from which it is evident that release of phosphate does occur very efficiently for the acetamido and carbamoyl derivatives; however, the large array of products of the phototrigger discouraged our further interest in these three electron-donating groups. The methoxy substituent (14c) showed a much cleaner behavior, yielding only two products from the chromophore, p-methoxyacetophenone and the rearrangement product p-methoxyphenylacetic acid. The p-hydroxyphenacyl phosphate (33) gave the rearranged p-hydroxyphenylacetic acid when photolyzed in mixed aqueous organic solvents (Eq. (69.12)); in fact, of all of the groups examined, only p-hydroxy and p-methoxy produced any rearranged phenylacetic acids. O HO OR O P OR O 33, R = Et OH hn, H2O O HO + HO OR P OR O 34 (69.12) The initial discovery that diethyl p-hydroxyphenacyl phosphate exclusively followed a rearrangement pathway was followed by an extension of our study to p-hydroxyphenacyl ATP (35). Irradiation of 35 at 350 nm released ATP and p-hydroxyphenylacetic acid with a quantum efficiency of 0.37 ± 0.01 and a rate constant for ATP appearance of 5.5 ± 1.0 × 108 s–1 [Eq. (69.13)]. 1348_C69.fm Page 13 Monday, October 13, 2003 3:22 PM 69-13 Photoremovable Protecting Groups NH2 O O O O N OPOPOPO -O -O -O O N NH2 N N hn HO - 34 O ATP . = 0.37 OH OH 35 N O O O HO POPOP O -O -O -O N N N OH OH kr = 5.5 x 108 s-1 (69.13) While the mechanism of this process is unknown, the p-hydroxyphenacyl photorelease likely involves an initial triplet state deprotonation of the phenolic hydrogen. In one scenario, the initially generated triplet intermediate partitions between loss of a proton and C-O bond cleavage, as pictured in Scheme 4. The exact course of the reaction depends greatly on the leaving group, solvent, and substituents attached to the chromophore. Here, the triplet phenol undergoes the equivalent of a homolytic cleavage of the bond to the substrate. In this scenario, it was envisaged that initial homolysis of the C-Y bond might be followed by a rapid single-electron transfer process; that is, the triplet phenol is essentially converted to its conjugate base before other competing processes for the radical pair can intervene. In this sequence, a spirodienedione is eventually generated by electrocyclic closure of the intermediate zwitterion or possibly the diradical. The conjugate base formed by the proton loss undergoes bond reorganization to the putative spirodienedione 30 accompanied by release of ATP. Further hydration of the spirodienedione and bond reorganization lead to the phenylacetic acid that is suggested for both pathways. A second mechanistic scenario involves proton loss concomitant with direct neighboring group assistance for the release of the substrate and formation of the spirodienedione. The subsequent proposed nucleophilic hydrolysis of the spirodienedione follows as above. Current evidence from subsequent solvent and substituent studies favor the latter mechanism (vide infra). O HO 3 O Y hν, 300 nm Y -H ST HO Y = (R'O)2PO2- khet khom HO +Y Y O pKa3 O 3 O + -Y H2O ket O O 29 kH 28 34 SCHEME 4 Proposed triplet state mechanisms for photorelease of substrates from the p-hydroxyphenacyl protecting group. The onset of the triplet-state phosphorescence emission of several p-hydroxyphenacyl esters indicated triplet energies of 68.9 to 70.6 kcal/mol. The phosphorescence emissions were quenched by sodium 2naphthalenesulfonate or potassium sorbate. Quenching studies confirmed the reactivity of the triplet state and further provided a lifetime of 5.5 ns for the triplet with a release rate of 1.82 × 108 s–1 in later studies (vide infra). 1348_C69.fm Page 14 Monday, October 13, 2003 3:22 PM 69-14 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition 1 O O hn OAc OAc ESIPT (H2O)n HO HO 36 * HO O CH3 O O O O HO -HOAc H2O OH 37* O 34 30 * = singlet or reactive ground state intermediate SCHEME 5 ESIPT singlet state photorelease mechanism of p-hydroxyphenacyl acetate.37 HO 0.8 O – 39 O O pH 7.05 pH 5.32 pH 5.05 pH 4.66 pH 4.38 pH 4.04 pH 2 38 0.4 0.0 350 400 450 500 / nm FIGURE 69.1 Transient absorption spectra obtained by LFP of p-hydroxyacetophenone in water (10% CH3CN) with various buffers. (From Conrad II, P. G. et al., J. Am. Chem. Soc., 122, 9346–9347, 2000. With permission.) The original proposal that the triplet state was the reactive state was challenged by Zhang et al.37 An excited singlet state or possibly a tautomeric ground state (e.g., 37*) was proposed as the reaction intermediate. In their studies, quenching by triplet quentchers was not observed during photolysis of phydroxyphenacylacetate (36), suggesting that the release of acetate occurred through an excited singlet state possibly involving an intramolecular proton transfer. They postulated that the excited singlet state intramolecular proton transfer (ESIPT) mechanism would form the quinone methide 37* that could either continue on to the spirodienedione (30) or decay to ground state and subsequently undergo release of acetate to form 34. Such a mechanism has precedence in the earlier work of Wan37,38a and Yates.38b Laser flash photolysis (LFP) studies by Givens and Wirz39 with diethyl pHP phosphate (Eq. (69.12)) confirmed the intermediacy of the phenacyl triplet state. Energy transfer quenching to naphthalene gave a rate of formation of the naphthalene triplet of 7.8 × 109 M–1 s–1. The presence of dioxygen increased the decay rate of the pHP phosphate triplet (kq ≈ 3 × 109 M–1 s–1). It was estimated from this study that pHP intersystem crosses with a rate constant of 3.1 × 1011 s–1 in aqueous acetonitrile. Quenching studies of the photochemical release of substrates from a series of pHP derivatives employing potassium sorbate gave excellent linear Stern–Volmer quenching results with lifetimes of 10–8 to 10–9 s for their pHP triplet states. These combined results firmly established the triplet as the reactive excited state. LFP studies on the parent chromophore proved revealing. The p-hydroxyacetophenone triplet undergoes a facile adiabatic proton tautomerization converting the phenol 28 into its conjugate base 38. 1348_C69.fm Page 15 Monday, October 13, 2003 3:22 PM 69-15 Photoremovable Protecting Groups O OH pKE = 16.4 (-1.0) O HO 39 28 pKa = -8.5 (4.6) pKa = 7.9 (3.6) O O 38 SCHEME 6 Tautomerization of p-hydroxyacetophenone in the ground state and triplet excited state (in parentheses). (From Conrad II, P.G. et al., J. Am. Chem. Soc., 122, 9346-9347, 2000. With permission.) Evidence for this unusual adiabatic proton transfer to solvent from the triplet p-hydroxyacetophenone came from analysis of the transient triplet absorption spectra of 28 as a function of the pH (Figure 69.1). These results were further supported by DFT calculations that provided the pKE values for the equi39 in their ground and triplet excited states. A pKa3 for 283 of 3.6 was derived from librium 28 these data and the triplet state pKa3 of 39 (4.6) and the known ground state pKa of 28 (7.9) (Scheme 6). These studies revealed an increase in acidity of over four pKa units relative to the ground state. The proton tautomer 39 is a non-productive intermediate because it is thermodynamically incapable of cleaving the ester C–O bond. Incorporating the rapid deprotonation that results from the large adiabatic decrease in pKa of 28 as a feature of the p-hydroxyphenacyl mechanism suggests that the conjugate base 38 is an attractive precursor to the rate-limiting release of the substrate. Because the triplet is formed with a ST rate constant of 3.1 × 1011 s–1, it is unlikely that there is any singlet state contribution to the deprotonation step. Rather, this appears to be exclusively a triplet process occurring on the excited triplet surface; that is, the two protonated species and the unprotonated ion undergo adiabatic proton tautomerizations (Scheme 6). The electron-rich aromatic ring increases the potential for intramolecular neighboring group attack at the α-carbon, leading to the release of the substrate and rearrangement of the chromophore. Thus, it becomes prudent to carefully explore the change in the pKa of the phenolic protons transitioning between the ground and excited triplet states as a key element in understanding the role played by aryl participation in the release step. Coumaryl and Arylmethyl Zimmerman’s early studies40 on the photosolvolysis of benzyl acetates in 50% aqueous dioxane set the stage for a variety of studies that employ m,m′-dimethoxybenzyl as a photoremovable protecting group (69.14). In general, the photofragmentation reactions of benzyl acetates are quite rapid, with rate constants of 108 s–1 or higher and are primarily singlet-state processes. According to Zimmerman, meta activation of the excited singlet state of benzyl acetates occurs through the approach of the excited- and ground-state energy surfaces, funneling the excited state toward heterolysis of the benzyl–ester bond. Substituents, including electron donors, in the para position lead primarily to homolytic fission and radical derived products. Direct heterolytic fission of the substrate-photoprotecting group bond is the required course for photorelease of most biologically important substrates. This process avoids the generation of destructive radicals that could result in reactions such as decarboxylation, radical dimerization, or redox processes. Thus, the effect of m-substitution on the photochemistry of benzyl, naphthyl, and other aromatic chromophores has become the object of many studies in search of alternatives to the o-nitrobenzyl class of protecting group. 1348_C69.fm Page 16 Monday, October 13, 2003 3:22 PM 69-16 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition -CO2 hn X hn 40 H3COCO SCHEME 7 Mechanistic scheme for arylmethyl ester photolysis in methanol.43 OCOCH3 hn , 50% aq. dioxane OCH3 H3CO OH . = 0.10 H3CO 40 OCH3 41, 79% (69.14) 41 42,43 on the nature of the meta effect Recent studies by both Zimmerman and DeCosta and Pincock have raised concerns about whether heterolytic or homolytic cleavage should be considered the primary photochemical process. According to Pincock, the mechanistic pathway for all substituted arylmethyl substrates begins with homolysis of the C–O ester bond to the substrate followed by a competition between electron transfer to an ion pair or typical ground-state radical reactions (Scheme 7).43 For those arylmethyl derivatives substituted with a meta electron-donating group, such as methoxy, the electron transfer occurs more rapidly than competing radical processes due to favorable redox properties of the radical pair. Normally, the details of the steps leading to the ion pair would not play a significant role in the outcome of the photorelease process except in those circumstances where the radical pair precursors have rapid, favorable divergent pathways available. Such could be the case for carboxylate esters, such as C-protected amino acid, peptides, and protein derivatives, where decarboxylation of the initially generated carboxy radical may compete with electron transfer to the ion pair. This deleterious process can become significant, leading to destruction of a portion of the released substrate. By either of these mechanisms, however, the productdetermining process for meta- and especially the di-meta-substituted arylmethyl chromophores leads principally to an ion pair, an intermediate arylmethyl carbocation, and the conjugate base of the leaving group. The coumaryl chromophore is essentially another arylmethyl analog, which has the attractive feature of high yields of fluorescence emission, sometimes a useful property for following the course of substratechromophore processes. One of the earliest studies using coumarin as a chromophore was the photorelease of diethyl phosphate from coumarylmethyl diethyl phosphate.26 The resulting coumarylmethyl cation covalently attaches to a wide variety of nucleophiles, as shown in Eq. (69.15): O OP(OEt)2 Nu hν, 360 nm Nu H3CO O O H3CO O O 42 43 Nu = CH3OH, piperidine, cysteine, tyrosine, α-chymotrypsin, HMT (69.15) Furuta et al.44 have reported the application of the coumaryl chromophore as a phototrigger for the release of cAMP (Eq. (69.16)); as shown in Table 69.9, the methoxy and hydroxy methylcoumarins gave the best conversions. 1348_C69.fm Page 17 Monday, October 13, 2003 3:22 PM 69-17 Photoremovable Protecting Groups TABLE 69.9 Percent Conversion of Coumarylmethyl cAMP After a 10 s Irradiation at 334 to 365 nm Caged cAMP R = Acetyl R = Propionyl R = Hydroxy R = Methoxy 23 9 64 60 Conversion (%) NH2 N N O O P O RO O NH2 N N N O O N OH O hν, 334 - 365 nm O HO N N O P O O OH (69.16) 44 The historical and mechanistic background for the most common photoprotecting groups were presented above. Examples that employ photoremovable protecting groups are given to illustrate the range and variety of the applications in chemistry and biology. As noted earlier, these are a very limited set of examples of the numerous published applications in biology and, to a lesser extent, in chemistry. Further information can be obtained from the reviews listed in References 1 through 7. 69.3 Carboxylic Acids o-Nitrobenzyl Because o-nitrobenzyl derivatives have been the most widely applied photoremovable protecting groups, modifications of this chromophore have received considerable attention. A recent study45 employing 2,2′dinitrobenzhydryl (DNB) for N-methyl-D-aspartate (NMDA) probed the NMDA receptor, which is one of the general classes of known glutamate receptors. The carboxyl group of NMDA was esterified with DNB, a stronger UV absorber (λmax 350 nm: ε = 1.69 × 104 M–1 cm–1) than typical o-nitrobenzyl analogs. The poor aqueous solubility of DNB-NMDA, however, required addition of 20% DMSO to attain complete dissolution. A single 308-nm laser pulse was sufficient to release NMDA within 4.2 µs, with a quantum efficiency of 0.18, as shown in Eq. (69.17). O2N O O hν, 308 nm O HO H3C NH O NO2 pH 7 O NO2 + NO OH HO H3C NH O NMDA β-O-DNB-NMDA (69.17) The time constant for the release of NMDA is pH dependent, occurring within 3.8 µs at pH 3.8 and 13.8 µs at pH 10.6, consistent with the rate of decay of the aci-nitro intermediate. The relatively rapid release rate from DNB suggests that this protecting group could be useful for further studies of the NMDA receptor. The versatility of the o-nitrobenzyl group has also been demonstrated in solid-phase synthesis. The phosphate group of the nucleotide tethered to a carboxylic acid through an alkyl chain provides a convenient link to the o-nitrobenzyl group, which is attached to a solid support. Synthetic manipulation of the oligonucleotide can be carried out under standard conditions and then release of the synthesized 1348_C69.fm Page 18 Monday, October 13, 2003 3:22 PM 69-18 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition TABLE 69.10 Isolated Yields of Completely Deprotected Oligonucleotides Protection (%) Deprotection (%) 45 Deprotection (%) 45′′ 96 89 97 90 70 91 92 69 77 80 71 70 a b c d oligonucleotide from the support is conducted photochemically.46 The carboxylate and substituted onitrobenzyl alcohol were coupled in 89 to 97% yields followed by the controlled pore glass (CPG) loading. Standard oligonucleotide synthesis was then carried out at the 3′-terminus to obtain 45a–d and 45′′a–d (Eq. (69.18). DMTrO 1. std. oligonuceotide synthesis 2. hn, 400 nm O n NO2 O H3CO HO H N O O P O (CH2)n+1CO2H O Oligo. 3. detritylation 4. NH4OH n 1 2 3 4 a b c d O 45a-d (n = 1-4) Oligo = T20 45'a-d (n = 1-4) Oligo = TAC GCA ATC CTA GAT CTA AT DMTr = 4,4'-dimethoxytrityl (69.18) Upon completion of the synthesis, the oligonucleotides were severed from the CPG solid support photochemically using a 400-nm light source. The efficiency of the release process was as high as 92%, as shown in Table 69.10. This protocol is useful for elaborating the 3′-terminus of oligopeptides using mild, traceless reagent conditions at room temperature and neutral pH. Coumaryl The photochemical and photophysical behavior of 4-(hydroxymethyl)-7-methoxycoumarin (MCM) caged acids was studied under physiological conditions by Bendig et al.47 (Eq. (69.19)). O OH X hn, 333 nm MeO O + MeO O O HOX O R CH3 X= O O 46a 46b: R = OCH3 46c: R = H 46d: R = CN (69.19) Photocleavage of the excited singlet state of MCM caged compounds is thought to proceed via a photoSN1 mechanism (solvent-assisted photoheterolysis). Evidence favoring this mechanism was found from irradiation of MCM caged derivatives in 18O-labeled water, which exclusively incorporated the 18O-label in the MCM–18OH product (see Scheme 8). The deprotection process of the MCM derivatives was 1348_C69.fm Page 19 Monday, October 13, 2003 3:22 PM 69-19 Photoremovable Protecting Groups TABLE 69.11 Data for 4-(Hydroxymethyl)-7-Methoxycoumarin (MCM) Caged Acids Acid Protection (% Yield) Deprotection (Φ) 46a 46b 46c 46d 82.6 90.0 98.0 83.8 0.0043 0.0045 0.0052 0.0064 CH3 1 MC CH2 OX * 1 MC CH2 OX * R-H H3CO kset -kset hn O O (trace) k1 MC CH2 OX k0 kesc MC CH2 OX MC = 7-methoxycoumarin-4-yl moiety MC CH2 OH (solvent) MC CH2 OH + OX H (solvent) HOX SCHEME 8 Reaction pathways for photolytic cleavage of MCM esters. dependent on the leaving group and the solvent polarity. The quantum efficiencies for the reactions of 46a–d were low, in the range of 0.0043 to 0.0064 as listed in Table 69.11. MCM–OH is a highly fluorescent product that can be conveniently monitored during the course of the reaction. Furthermore, MCM caged compounds are very stable to the hydrolysis. Phenacyl The rapid release (~108 s–1) of substrates from the p-hydroxyphenacyl (pHP) group enables fast biological processes to be studied. p-Hydroxyphenylacetic acid (34) is generated with a quantum efficiency (Φrea) of ~0.18. In contrast, the presence of added electron-donating substituents on the aromatic ring of the pHP group makes the rearrangement a minor pathway for 47′′a–b and completely suppress it for 47′′′a–b (Scheme 9).48 The quantum efficiencies for the disappearance of the various pHP esters, the appearance of (34), and corresponding pHP-protected substrates are given in Table 69.12 . A complication that could arise in such systems is the potential for decarboxylation of the released carboxylate ion. However, no decarboxylation products were observed within the detection limits of 1HNMR and HPLC. With the production of a biologically benign photoproduct, the pHP protecting group has proven to be an efficient tool for investigations of fast biological processes. For example, Givens et al.49 applied the pHP phototrigger to the investigation of the bradykinin BK2 receptor. It is known that bradykinin acts as an active pain-transducer when released during tissue damage. A major difficulty in studying the detailed physiological mechanism of the action of bradykinin is a concomitant rapid enzymatic degradation of the nonapeptide immediately after its release from its precursor protein. Therefore, the photorelease from pHP bradykinin (48), which protects bradykinin from degradation of the agonist prior 1348_C69.fm Page 20 Monday, October 13, 2003 3:22 PM 69-20 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition TABLE 69.12 Quantum Yields of pHP Esters for Irradiation at 300 nma Ester 47a 47b 47c 47d 47e 47f 48 Solvent (Water/AcCN) Φdis Φapp Φrea Water Water 6:4 6:4 6:4 7:3 Water 0.21 0.14 ~0.2b 0.18 ~0.18b 0.24 0.21 0.21 0.14 — 0.17 — 0.23 0.22 0.19 0.08 — 0.14 — 0.17 0.19 pHP GABA pHP glutamate pHP cyclopropylacetate pHP phenylacetate pHP pivalate pHP oleate pHP bradykinin An NMR tube was charged with ~10 mg (~40 µmol) of the appropriate photoprotected acid and 10 mol% of 1,2,3-benzenetricarboxylic acid as an internal standard in 2 mL of solvent. The quantum efficiencies were determined at less than 20% conversion of the starting ester. By comparison with 47d. a b Abbreviations: dis = disappearance, app = appearance of acid, rea = rearrangement to phenylacetic acid. O HO 3 O 1. hn 2. ISC 3. deprotonation OCOR' OCOR' O R 47a-f 47 R = H 47' R = 3-OMe 47" R = 3,5-OMe R 47* reduction rearrangement H2O O O HO OH + OH HO R + R'CO2 O HO O R R O R SCHEME 9 Photoreaction for the pHP esters. to release, facilitates the investigation of its action during the transduction process by allowing precise temporal and spatial release of bradykinin to activate the bradykinin BK2 receptor (Eq. (69.20)). O NH H2N N H N N O O N H H N O HO O N H N O O N H NH2 H2N 48 H N O NH O CO O OH N H hn, 337 or 300 nm NH2-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg-CO2 + 34 D2O, Pyrex (69.20) pHP Bradykinin 48 was obtained in an overall yield of 84% by derivatizing the partially protected bradykinin, obtained by cleavage of the C-terminus from the resin after sold-phase Merrifield synthesis.49 1348_C69.fm Page 21 Monday, October 13, 2003 3:22 PM 69-21 Photoremovable Protecting Groups Reaction of the C-termnus with p-hydroxyphenacyl bromide followed by treatment with 1% TFA gave pHP bradykinin 48, free of the other protecting groups employed during the solid phase synthesis.49 A single 337-nm flash (<1 ns) released sufficient bradykinin to excite the BK2 receptors on single rat sensory neurons, which dramatically increased the intracellular calcium concentration measured with Indo-1, a Ca2+-chelating fluorescent indicator. The quantum efficiency of bradykinin appearance was independently determined to be 0.22, as shown in Table 69.12. Benzoin There are a number of examples of benzoin and substituted benzoin esters that have been employed as photoremovable protecting groups for carboxylic acids. Among these, the application of benzoin as a traceless linker for solid-phase synthesis of oligopeptides and introduction of Fmoc-protected amino acids by Balasubramanian50 is instructive. As an example, the release of Fmoc-Ala shown in Eq. 69.21 occurs upon photolysis at 350. Balasubramanian found that the maximum yield was obtained after a 2h photolysis as determined by HPLC. NHFmoc HO O O hν, 350 nm O Ph + NHFmoc O O Ph O O (69.21) In order to avoid premature photolysis of the benzoin linker by adventitious room light during the course of the synthesis, the dithiane-protected 3-alkoxybenzoin (49a) has been suggested as a “UV-inactive” linker. Dithiane 49 released less than 3% of the product after irradiation at 350 nm. The photosensitivity is restored by hydrolysis of the dithiane prior to photocleavage, as illustrated in Eq. (69.22). Photolysis of the deprotected linker resulted in a 75% yield of the product. O NHFmoc O S S Ph (i) NHFmoc O O O O Ph O 49b 49a (i) (a) bis[(trifluoroacetoxy)iodo]benzene, (b) mercury (II) perchlorate, or (c) periodic acid (4 eq.), THF:water (10:1) (for a and c), THF (for b), ambient temperature, 18 h. >95% conversion (69.22) Other A series of 2,5-dimethylphenacyl (DMP) esters were photolyzed in benzene or methanol (Eq. (69.23)). O R O O 50 hν, >300 nm O O + benzene or methanol HO R 85-95% 51 (69.23) 1348_C69.fm Page 22 Monday, October 13, 2003 3:22 PM 69-22 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition TABLE 69.13 Data for 2,5-Dimethylphenacyl Esters Protection (%Yield) R CH3 C 6H 5 C 6H 5 C6H5CH2 n-Pentadecyl N-(t-butoxycarbonyl)-L-phenylalanine a b Photolysis Condition (nm) 84 95 95 84 69 51 Deprotection (% Yield)a 85b 86 92 91 95 90 Benzene, >280 Benzene, >280 Methanol, >254 Benzene, >280 Benzene, >280 Benzene, >280 Isolated yield of the crude acids (>95% purity). Determined by gas chromatography. TABLE 69.14 Quantum Yieldsa of DMP Esters Protection (% Yield) X –OCOPh 95 –OCOCH2Ph 84 –OCOCH3 84 a Photolysis Condition Deprotection (Φ) Benzene Methanol Benzene Methanol Benzene Methanol 0.23 0.09 0.18 0.11 0.25 0.14 Quantum yield for ester release; valerophenone was used as an actinometer; irradiated at λ >300 nm. Error margins are approximately 10%. The formation of the corresponding carboxylic acids occurred with almost quantitative isolated yields, as shown in Table 69.13.51 In contrast with the structurally related p-hydroxyphenacyl esters, the release mechanism from 50 occurs through an efficient intramolecular γ-hydrogen abstraction via the (n,π*) excited ketone. MeO O X hν, >300 nm 51 + O + HX (69.24) Recently, Wirz and Klan52 reported LFP studies on several of the DMP esters in benzene and methanol (Eq. (69.24)). Quantum efficiencies (Φ) in benzene are 0.18 to 0.25, while those in methanol fell to 0.09 to 0.14. Thus, this photoprotecting group appears to be better suited to applications in nonpolar media such as benzene rather than methanol (Table 69.14). A novel design of “orthogonal” protecting groups, i.e., the removal of protecting groups selectively from a multi-protected substrate, has been reported by Bochet53 in which the irradiation wavelengths 1348_C69.fm Page 23 Monday, October 13, 2003 3:22 PM 69-23 Photoremovable Protecting Groups serve as the “orthogonal reagents”. A mixed diester of pimelic acid was capped with a dimethoxybenzoin group at one end and an o-nitrobenzyl group at the other terminus. Despite the possibility that intramolecular energy transfer or equilibration could occur between the two chromophores, selective photolysis led to the sequential removal of each with high chemical yield, as shown in Eq. (69.25). Upon photolysis of 52 at 254 nm for 5 minutes, 92% of 53b was released, whereas irradiation at 420 nm for 24 hours released 70% of 53a, as determined by 1H NMR. O O MeO O NO2 OMe 52 1. hν, 420 nm 2. TMSCHN2 70% O O O 1. hν , 254 nm 2. TMSCHN2 92% MeO MeO OMe O OMe 53a O O Ph O MeO OMe O O MeO Ph NO2 (69.25) 53b OMe 69.4 Phosphates and Phosphites o-Nitrobenzyl The most notable example of o-nitrobenzyl (ONB) caged phosphates remains that reported by Trentham and co-workers54 on the synthesis and photochemistry of caged ATP in the late 1980s. Caged ATP, P3-1(2-nitrophenyl)ethyladenosine 5′-triphosphate (54), was synthesized in nearly quantitative yield in three steps starting with commercially available o-nitroacetophenone. Reaction with hydrazine to give the corresponding hydrazone, followed by oxidation with MnO2 provided the aryldiazoethane precursor that was used to alkylate ATP. The photolysis of caged ATP furnished ATP with an efficiency of 0.63, as illustrated in Eq. (69.26): NH2 N O O O O P O P O P O OOONO2 54 N O OH OH N N hν, 320 nm TES buffer, KCl, MgCl2, pH 7.1 Φ app = 0.63 ATP + O NO (69.26) The rate of release of ATP was found to be dependent on the pH and the relative concentration of magnesium ion in solution. Pelliccioli and Wirz5 have shown that the rate-determining step is the decay of the hemiacetal (or hemiketal) intermediate between pH 4 and 8. In this region, the slow hydrolysis of the hemiacetal limits the mechanistic value of the o-nitrobenzyl protecting group to studies of relatively slow reactions (e.g., kr < 103 s-1). The nitroso byproduct has also proved problematic for spectroscopic analyses of ONB reactions due to its reactivity with some substrates and with proteins. This problem was circumvented by conducting the photolysis in the presence of dithiothreitol, a hydrophilic thiol and an excellent nucleophile that readily sequesters the nitroso byproduct. 1348_C69.fm Page 24 Monday, October 13, 2003 3:22 PM 69-24 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition TABLE 69.15 Results of Photolysis of cAMP and cGMP Coumaryl Esters Coumaryl Derivative 55a 55b 55c a Φdisa λmax (nm) 0.21 (0.25) 0.12 (0.16) 0.10 (0.14) 402 326 346 Solvent 80:20 HEPES-KCl buffer/MeOH HEPES-KCl buffer, pH 7.2 HEPES-KCl buffer, pH 7.4 For the cAMP ester; quantum efficiency for the cGMP derivative is in parentheses. Coumaryl Coumarylmethyl esters have been used as photoprotecting groups by several groups (e.g., Furuta et al.55,56 and Hagen et al.57) for deprotection of cAMP and cGMP. To overcome the limited aqueous solubility of these derivatives, Hagen has modified the coumaryl chromophore with carboxyl and amino substituents.57 Three new variants of the coumaryl system were synthesized and their photochemistry explored — (7diethylaminocoumarin-4-yl)methyl (DEACM), (7-carboxymethoxycoumarin-4-yl)methyl (CMCM), and (6,7-bis[carboxymethoxy]coumarin-4-yl)methyl (BCMCM) as esters of cAMP and cGMP. Photolysis resulted in liberation of the free cyclic phosphate along with the hydrolyzed chromophores, i.e., 56a-c (Eq. (69.27)). R1 R1 R2 R2 hν, 333 nm O A (or G) O O O P O O O HO cAMP or cGMP + OH O O 56 55a R1 = H, R2 = Et2N 55b R1 = H, R2 = OCH2CO2H 55c R1 = R2 = OCH2CO2H (69.27) The caged coumaryl compounds were synthesized in 11 to 34% yield, clearly a limitation with this photoremovable protecting group. The addition of the carboxymethoxy groups in 55b and 55c dramatically enhanced the water solubility of these analogs as compared with the ester 55a, and their photorelease occurred with good quantum efficiencies as seen in Table 69.15. On the other hand, the less water-soluble 55a had the best quantum efficiency, and its absorption maximum was the most red shifted in the series. Phenacyl In light of its inherent advantages over other chromophores, the p-hydroxyphenacyl group has received recent attention as a promising photoprotecting group for phosphates. Recent reports on p-hydroxyphenacyl esters of phosphate, diethyl phosphate, and ATP by Givens et al.36 and on GTP by Du et al.58 indicate that these phosphate esters undergo efficient photorelease of the phosphoric acid moiety along with the rearranged p-hydroxyphenylacetic acid as the sole photoproducts of the reaction (Eq. (69.28)). 1348_C69.fm Page 25 Monday, October 13, 2003 3:22 PM 69-25 Photoremovable Protecting Groups O O OP OR1 OR2 HO O hν, 300 nm - 10% CH3CN/TRIS buffer, pH 7.3 O-P OR1 OR2 + 34 57a R1 = R2 = Et 57b R1 = R2 = NH4+ 57c R1 = GDP, R2 = NH4+ 35 R1 = ADP, R2 = NH4+ (69.28) TABLE 69.16 Data for pHP Caged Phosphates Caged Phosphatea Protection (% Yield) Deprotection (% Yield) Φdis 57a 57b 35 57c 87 96 42 20 Not determined Quant Quant Not determined 0.77 0.38 0.37 (0.30)b Not determined a b Results shown for 57c are from Du et al.;58 derivative 57c was photolyzed at 308 nm. The value in parentheses for 35 is the quantum efficiency for the appearance of ATP. Derivative 57a was synthesized by reacting the phosphate directly with p-hydroxyphenacyl bromide. Derivative 57b was synthesized from hydrogenolysis of the pHP dibenzyl phosphate ester. For 35 and 57c, the pHP monophosphate (57b) was first protected as the corresponding ketal that was then coupled with ADP or GDP, respectively. The caged ATP and GTP analogs were then obtained by hydrolysis of the ketal. Yields and quantum efficiencies for the disappearance of the pHP phosphate esters are given in Table 69.16. pHP caged ATP was recently used as a probe in the study of Na,+ K+-ATPase, an enzyme involved in the intracellular transport of sodium and potassium ions.59 Membrane samples possessing the ionchannel proteins were bathed in caged ATP which was activated by UV laser flash photolysis. Na,+ K+ channel transport was observed as a result of the activation of the enzyme by the released ATP. Direct spectroscopic evidence of the release of ATP was obtained by time-resolved Fourier transform infrared (FTIR) spectroscopy (Figure 69.2). The changes in the characteristic absorptions of the prominent functional groups of the reactant and product include the disappearance of the γ-PO2– ester band of the caged ATP at 1270 cm–1 and the appearance of the free γ-PO3–2 band of the released ATP at 1129 cm–1. Benzoin In 1994, Pirrung and Shuey60 reported the protection of phosphates using dimethoxybenzoin. Resolved (R)3′,5′-dimethoxybenzoin (optically active) was converted to the phosphoramidite by treatment with diisopropylaminocyanoethoxychlorophosphine. Subsequent reaction with the appropriate primary or secondary alcohol followed by oxidation led to the series of phosphate esters 58a-e in moderate to high yields: 1348_C69.fm Page 26 Monday, October 13, 2003 3:22 PM 69-26 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition 0.015 CO2-of product ATP PO32- ∆Abs. 0.010 0.005 0.000 -0.005 Caged ATP PO2- -0.010 2000 1900 1800 1700 1600 1500 1400 1300 1200 1100 1000 900 Wavenumber / 1/cm FIGURE 69.2 Time-resolved FTIR difference spectrum for the photolysis of pHP caged ATP. The absorbance was measured 10 ms to 11 s after the photolysis flash and subtracted from the absorbance prior to photolysis. (We thank Professors Klaus Fendler and Andreas Barth for the TR-FTIR results with pHP ATP.) MeO O CH2CH2CN O P OR O Ph hν, 350 nm O O O P OR + CN -O 11 OMe 58 CO2Me CO2Me R= NHBoc NHBoc Me Me TrO O O T Me Me O O O 58a 58b 58c 58d 58e (69.29) Photolysis of 58a-e shown in Eq. (69.29) led to release of the phosphate derivative along with the benzofuran byproduct 11. The overall sequence from the phosphoramidites to phosphate esters 58a–e was accomplished in moderate yields, and the unprotected phosphates were obtained in good yields upon photolysis, as seen from Table 69.17. TABLE 69.17 Yields for the Photolysis of DMB Phosphoramidites R Protection (% Yield) Deprotection (% Yield) 58a 58b 58ca 58d 58e 45 82 58 60 55 85 86 85 83 87 a The (R) enantiomer. 1348_C69.fm Page 27 Monday, October 13, 2003 3:22 PM 69-27 Photoremovable Protecting Groups 69.5 Sulfates and Other Acids Sulfonic acids have largely remained unexplored in terms of a functional group that is released from a photoremovable protecting group. One recent example has been reported by Bendig et al.,47 in which methanesulfonic acid was protected as the corresponding methoxycoumarin derivative. (7-Methoxycoumarin-4-yl)methylmethanesulfonate 59 was synthesized from reaction of methanesulfonic acid with 4(diazomethyl)-7-methoxycoumarin in refluxing chloroform. Photolysis at 333 nm resulted in the release of methanesulfonic acid, as shown in Eq. (69.30). O S CH3 O O OH hν, 333 nm + H3CO O O 7:3 HEPES buffer/MeOH pH 7.2 O H3CO 59 O O HO S CH3 O 60 HEPES = N-(2-hydroxyethyl)piperazine-N'-(2-ethanesulfonic acid) (69.30) While the protection yield for 59 was low (26%), the quantum efficiency of 0.081 is reasonable when compared with other leaving groups that were reported in this study. The chemical yield of the deprotection was not provided; however, the photoproduct 60 was recovered in ≥95% yield, suggesting a high yield of the released sulfonic acid. 69.6 Alcohols, Thiols, and N-Oxides o-Nitrobenzyl Among the most common photoprotecting groups used for alcohols are the o-nitrobenzyl derivatives. Recently, Iwamura61 reported the synthesis and photochemistry of caged carbohydrates protected with an o-nitrobenzyl group at one or more positions within the molecule. Equation (69.31) illustrates a typical example in which photolysis of the caged glucopyranoside 61 at 350 nm in methanol led to the release of the free methylglycoside 62 in 60% yield, along with the nitroso byproduct 3. NO2 O HO HO OH O hν, 350 nm MeOH OH O HO HO HO + OMe OMe 61 3 62 (69.31) Derivative 61 was synthesized in 71% yield by reductive bond cleavage of the corresponding 4,6-O-onitrobenzylidene acetal of methyl 3,4-acetyl-β-glucoside with triethylsilane and boron trifluoride etherate, followed by deacetylation of the 3,4-diacetates with sodium methoxide in methanol. o-Nitrobenzyl chemistry was also extended to the release of thiols as demonstrated by Smith et al.,62 who demonstrated that a cysteine congener was protected as the corresponding thioether 63 in 89% yield. Photocleavage occurred at 366 nm to give the liberated Boc-protected thiol 64 in 44% yield in the presence of semicarbazide hydrochloride, a carbonyl scavenger (Eq. (69.32)). 1348_C69.fm Page 28 Monday, October 13, 2003 3:22 PM 69-28 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition O O NHBoc H2N S hν, 366 nm NO2 NHBoc H2N + 1:1 CH3CN/0.05 M PBS 3 SH pH 6 64 63 (69.32) Finally, the addition of ascorbic acid as an antioxidant to the reaction mixture gave a quantitative yield of 64. No thiol was recovered when the photolysis was carried out in the absence of ascorbic acid, despite experimental evidence of the disappearance of 63. While the o-nitrobenzyl system is well accepted as a photoremovable protecting group, it nevertheless suffers the limitation of toxicity to the biological entity and a highly reactive nitroso functional group formed as the byproduct. As was seen in the case of the cysteine cogener 63, the photodeprotection may require a scavenging or reducing agent if thiols are to be generated in such a strategy. In the late 1990s, Pfleiderer and co-workers63 developed a new, β-substituted variant of the o-nitrobenzyl chromophore, 2-(o-nitrophenyl)ethoxycarbonate. The 5′-O-2-(o-nitrophenyl)ethoxycarbonyl thymidines were obtained from 2-(o-nitrophenyl)ethanol carbonates by reaction with diphosgene under basic conditions, followed by treatment with thymidine in anhydrous methylene chloride at reduced temperature. The synthetic yields ranged from 41 to 81%. Photolysis of a 0.1-mM solution of the photoprotected thymidine 65 in a 1:1 methanol/water mixture at 365 nm resulted in the release of thymidine (66), carbon dioxide, and the photolabile o-nitrostyrene derivative 67 (Eq. (69.33)).The photorelease is believed to occur through a β-elimination mechanism64,65 from the aci-nitro intermediate 67′′ shown in Scheme 10. NO2 hν O O OH N O NO2 O O OR' + R R R R'OH -CO2 OR' 67' 67 SCHEME 10 β-Elimination mechanism proposed for 2-(o-nitrophenyl)ethoxycarbonyl thymidines. O O HN R3 HN NO2 R2 O O R1 O O R N hν, 365 nm O 1:1 CH3OH/H2O O HO O R3 N + -CO2 NO2 R2 R1 R OH OH 66 65 65a 65b 65c 65d R = CH3, R1, R2,R3 = H R = o-nitrophenyl, R1, R2,R3 = H R = H, R1 = I, R2,R3 = H R = H, R1 = Br, R2,R3 = H 65e 65f 65g 65h 67 R = H, R1 = Cl, R2,R3 = H R = H, R1, R3 = Cl, R2 = H R = H, R1 = F, R2,R3 = H R = R1 = H, R2,R3 = OCH3 (69.33) 1348_C69.fm Page 29 Monday, October 13, 2003 3:22 PM 69-29 Photoremovable Protecting Groups TABLE 69.18 Yields for 2-(o-Nitrophenyl)ethoxycarbonyl Thymidine Derivatives Derivative Protection (% Yield) Φ Deprotectiona (% Yield) 65a 65b 65c 65d 65e 65f 65g 65h 71 70 68 73 80 81 70 41 0.35 0.20 0.10 0.076 0.070 0.072 0.037 0.0013 76 nd nd nd 80 nd nd nd a Based on the recovery of thymidine; Buhler, S., Giegrich, H., and Pfleiderer, W., Nucleosides & Nucleotides, 18, 1281-1283, 1999. nd = not determined Some advantages of this nitrobenzyl variant include the lack of the nitroso byproduct and release rates that are relatively fast compared with the parent o-nitrobenzyloxycarbonyl derivative. For example, the release of thymidine from 65b is reported to be twice as fast as that for the α-substituted derivative; however, the nitrostyrenes (i.e., 67) are photolabile and thus can compete with the starting material for incident light. The quantum efficiencies for the release of thymidine varied depending on the substituents on 65. Substitutions at the benzylic carbon appeared to give the highest release efficiencies, whereas the chloro derivatives gave slightly better conversions, as seen in Table 69.18. The high quantum efficiency and respectable protection and deprotection yields obtained for the methyl-substituted 2-(o-nitrophenyl)ethoxycarbonyl photoprotecting group prompted Pirrung et al.66 to use this protecting group for the solid-phase synthesis of oligodeoxynucleotides. Thiopixyl and Coumaryl Coleman and Boyd67 introduced the 9-phenylthioxanthyl or S-pixyl photoprotecting group for the four principal nucleosides, thymidine and three other benzoyl protected nucleoside bases. The chromophore was synthesized in three steps, starting with thioxanthone, through Grignard addition of phenylmagnesium bromide, followed by dehydration with trimethylsilyl chloride and dimethylsulfoxide to give the 9chloro-9-phenylthioxanthene. Treatment with the corresponding alcohol in a dry solution of pyridine and dimethylaminopyridine (DMAP) afforded the photoremovable protected hydroxy derivatives 68 in good yields (Table 69.19). Irradiation of 68 in aqueous acetonitrile resulted in the release of the nucleoside or alcohol along with 69 (Eq. (69.34)). Ph OR Ph OH hν, 300 nm S + CH3CN(aq) ROH S 68 69 X CH2 R= CH2 O CH2 OH X = Thymine, N-Bz-Adenine, N-Bz-Cytosine, N-iBu-Guanine (69.34) 1348_C69.fm Page 30 Monday, October 13, 2003 3:22 PM 69-30 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition TABLE 69.19 Synthesis and Deprotection Yields for Various Derivatives of 68 R Protectiona (% Yield) Deprotectionb (% Yield) 80 94 79 86 82 92 93 95 97 96 75 89 CH2-benzyl CH2-(E)styryl Thymidine N-Bz-adenosine N-Bz-cytidine N-iBu-guanosine a b Based on isolated yields after column chromatography. Substrate concentrations were approximately 0.1 mM; yields were determined by HPLC. Mechanistically, this photorelease reaction occurs via photosolvolysis of the aryl ether carbon–oxygen bond. The resulting resonance stabilized S-pixyl carbocation reacts with water to form 69. Control experiments showed that the photoprotected alcohols are stable under thermal conditions; that is, refluxing in aqueous acetonitrile resulted in no detectable decomposition. Each of the caged products was obtained as a solid, a convenience for laboratory purification and manipulation. The best deprotection yields were obtained with solvent mixtures containing the maximum concentration of water permissible, limited only by the solubility of the protected alcohol. Concentrations of water ranged from 40 to 60%, depending on the type of alcohol used. In most cases, excellent deprotection yields were obtained. A factor that limits the versatility of the S-pixyl group is the wavelength range required for photolysis. The range of excitation wavelengths from 200 to 300 nm overlaps with a number of functional groups that could compete with the incident light. For example, the pyrimidine base cytosine has substantial absorptivity at 300 nm. As a result, extended irradiation times were required to effect deprotection of the N-Bz-cytidine analog and a lower yield was obtained, as seen from Table 69.19. Coumaryl photoprotecting groups have also been used for the protection of alcohols. A recent study by Lin and Lawrence68 described the synthesis and photorelease of caged diols using a coumaryl acetal derivative 70 (Eq. (69.35)). H R O O H Br O hν, 348 nm H OH Br + HO O O 1:1 CH3OH/buffer HO pH 7.4 70 OPh SPh Φ = 0.0041 70b Φ = 0.0053 OH Me O Me 70a R O 71 Me R= O 70c Φ = 0.0051 N Me Ph 70d Φ = 0.0278 (69.35) Acetal 70 was prepared in a two-step sequence starting with oxidation of 6-bromo-7-hydroxy-4hydroxymethylcoumarin with manganese dioxide, followed by addition of the corresponding diol in 1348_C69.fm Page 31 Monday, October 13, 2003 3:22 PM 69-31 Photoremovable Protecting Groups anhydrous toluene. Protection yields ranged from 94% for 70a to 15% for 70d. Photolysis of 70 at 348 nm in a methanol/aqueous buffer solution afforded the free diol accompanied by the aldehyde photoproduct 71. The specific deprotection yields were not provided but in all cases were reported to exceed 75%. The photorelease mechanism is not well understood but is speculated to proceed through an intramolecular ion pair69 generated from photoheterolysis of the carbon oxygen bond, as shown in Scheme 11. Attack of a water molecule at the electrophilic carbon generates a hemiacetal that eliminates the alkoxy group to afford the diol and byproduct 71. 1 O 70 hν O R * R O O O HO O HO O HO R HO O O HO O HO Br O Br Br Br R H2O 71 O O R + OH HO O O SCHEME 11 Mechanism proposed for the photorelease of caged diols from 70. This particular coumaryl variant offers a unique advantage in essentially being able to protect both hydroxyl groups of a diol using only one equivalent of the protecting group. In addition, the derivatives in this study were shown to be stable to hydrolysis in aqueous solvents at neutral or basic pH. For example, incubation of compounds 70a–d in a 1:1 methanol/buffer solution (pH 7.4) resulted in no detectable degradation after a period of two weeks. Despite these advantages, the range of applications of the coumarin group is limited thus far to 1,2- and 1,4-diols (1,3-diols were found to be inert to photolysis). Like the onitrobenzyl group, the coumaryl group also has the disadvantage of producing a highly absorbing photoproduct. In addition, further studies need to be carried out in order to elucidate the mechanism of the photocleavage. Benzoin In 1995, Pirrung and Bradley70 reported the use of dimethoxybenzoin (DMB) carbonate to protect various alcohols, including the 5′-hydroxyl group of nucleosides. The DMB carbonate was synthesized in three steps, starting with methylation of carbonyldiimidazole with methyl triflate followed by addition of 2(3,5-dimethoxyphenyl)-2-hydroxy-1-phenylethanone to form a relatively stable activated acylating agent. Treatment with an alcohol under basic conditions in nitromethane furnished the protected alcohol 72 in yields that ranged from 42 to 95%. Irradiation of 72 at 350 nm resulted in release of the alcohol and formation of dimethoxybenzofuran 11 (Eq. (69.36)). A wide variety of alcohols, including a thiol, were explored in this protection/deprotection scheme. Excellent deprotection yields were obtained, as high as 98% for the protected thymidine derivative. The mechanism as discussed earlier is postulated to proceed through intramolecular cyclization followed by demotion to form a zwitterionic intermediate. Expulsion of the alcohol occurs either concomitantly with the release of carbon dioxide or by a stepwise decarboxylation of the initially released carbonate 73. 1348_C69.fm Page 32 Monday, October 13, 2003 3:22 PM 69-32 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition RO O O MeO Ph O hν, 350 nm O O RO THF OMe -CO2 (36) ROH 73 -11 72 PhCH2OH 88% PhCH2SH N-Bz-adenosine N-iBu-guanosine thymidine 95% 97% 96% 98% C8H17 OH HO 96% 94% (69.36) Attractive features of the DMB photoprotecting group are that the benzofuran photoproduct 11 is inert and exhibits strong fluorescence at 396 nm, allowing the deprotection conversions to be monitored spectroscopically. In the same study, the DMB group was successfully used to synthesize two trinucleotides bearing adjacent thymidine residues, demonstrating its potential in solid-phase DNA synthesis. Compared with nitrobenzyl photoprotecting groups, the rate of release of substrate is much faster for the DMB group70 (krelease ~ 108–109 s–1). The main disadvantages of the DMB group are the competition for incident light by the photoproducts and poor solubility in aqueous media. Other The anthraquinon-2-ylmethoxycarbonyl (Aqmoc) photoprotecting group is a relatively recent addition to the chromophores used as photoremovable groups. In a recent application for alcohols, Furuta et al.71 reported that the caged derivative could be synthesized in two steps from anthraquinonylmethanol by treatment with 4-nitrophenylchloroformate and DMAP followed by coupling to the desired alcohol with DMAP to provide 74 in good yields (Table 69.20). Photolysis of 74 in 50% aqueous tetrahydrofuran (THF) at 350 nm resulted in the release of the alcohol (Eq. (69.37)). TABLE 69.20 Yields and Quantum Efficienciesa for Aqmoc Derivatives Aqmoc Derivative 74a 74b a b Protection (% Yield) Φ Deprotection (% Yield) 76 86 0.1 Not determined 68 91b For the disappearance of starting material. Determined by HPLC. 1348_C69.fm Page 33 Monday, October 13, 2003 3:22 PM 69-33 Photoremovable Protecting Groups O O O OR hν, 350 nm ROH photoproducts + THF/H2O O NH2 74 N O R= CH2 O O N CH2 N N O O O OH OH 74a 74b (69.37) The photoproducts from the anthraquinone moiety were not fully characterized; however, in the case of 74a, an anthraquinon-2-ylmethanol tetrahydrofuranyl ether was isolated, probably the result of hydrogen atom abstraction from the solvent. Also, a small amount of bis(1,2,3,4-di-O-isopropylidene-D-galactopyranosyl) carbonate was obtained after photolysis of 74a, likely the result of attack of the released alcohol on the starting material. Little is known about the photorelease mechanism of the Aqmoc group. Stern–Volmer analysis showed that the triplet excited state is the reactive excited state that leads to release of the alcohol. The rate constant determined for 74a (4.6 × 106 s–1) is consistent with rapid release of the carbonate followed by a slower, rate-limiting loss of carbon dioxide to give the free alcohol. Additional studies are needed to confirm this mechanism. Recently, it was shown that nitro-substituted aryl carbamates could be used as photolabile protecting groups for alcohols.72 N-Methyl-N-(o-nitrophenyl)carbamate 75 (Eq. (69.38)) was synthesized in two steps, beginning with acylation of N-methyl-2-nitroaniline with phosgene followed by nucleophilic addition of the alcohol, either as an alkoxide or in the presence of DMAP and triethylamine. An alternative synthetic route involved the generation of the corresponding chloroformate from the alcohol and phosgene followed by nucleophilic addition of the nitroaniline. The carbamate derivatives 75a-d were synthesized in yields ranging from 58 to 94%. Photolysis of carbamate 75 at various wavelengths led to deprotection of the alcohol, which was accompanied by formation of nitrosoaniline 76 as a byproduct. Deprotection yields for all derivatives were not provided; however, the reported yields for 75c and 75d were 100% and 91%, respectively. Such high yields are a definite advantage in terms of both synthetic and biological applications. Another attractive feature is the solubility in ethanol and water, solvents suitable for biological studies. Despite these advantages, two main limitations are worthy of note. First, the carbamate cages are susceptible to hydrolysis, particularly in basic media,73 thus limiting their use to aqueous solvents with relatively neutral pH. Second, longer irradiation wavelengths were found to result in decreased deprotection yields; for example, when 75c was irradiated at 312 nm, a quantitative deprotection yield was obtained. The yield dropped to 76% when the photolysis was carried out at 365 nm. NO2 NO OR hν, 254-365 nm N O N ROH H + EtOH/H2O 76 75 O R= CH3 75a CH2Ph 75b I Ph N H 75c 75d N O CO2Me (69.38) 1348_C69.fm Page 34 Monday, October 13, 2003 3:22 PM 69-34 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition TABLE 69.21 Caged Silyl Derivatives and Their Corresponding Deprotection Yields in Methanol Silyl Ether Protection (% Yield) Deprotection (% Yield) 77a 77b 77c 77d 77e 77f 82 80 77 79 81 85 94a 89 90 92 86a 90 a Based on the yield of the byproduct 78. Silyl photoprotecting groups have recently been developed for primary and secondary alcohols.74 The silyl cage, (2-hydroxy-3-naphthylvinyl)-diisopropylsilyl ether 77, was synthesized in nine steps starting with the commercially available naphthalene-2,3-diol. Irradiation at 350 nm in methanol triggered the release of the alcohol, accompanied by the formation of a cyclic silyl byproduct 78 (Eq. (69.39)). This byproduct is likely formed via intramolecular attack of the naphthol oxygen at silicon following a trans,cis-isomerization of the starting material. Synthetic and photochemical yields are listed in Table 69.21. Si OR OH hν, 350 nm O CH3OH 77 R= i-Pr Si + 78 O t-Bu T DMT-O O T Me 77b 77c H3C CH3 OTBS 77a ROH 77d 77e CH3 77f (69.39) Byproduct 78 exhibits its strongest absorption in the region below 310 nm and, therefore, does not significantly compete with 77 for incident light. The yields from Table 69.21 are sufficiently high to enable the practical use of the silyl photoprotecting group in synthetic applications; however, the silyl cages lack the water solubility necessary for application in aqueous media. Like triisopropylsilyl ethers, the cages are also susceptible to cleavage in the presence of acidic media or solutions containing fluoride such as 1-N HCl or TBAF.75 The importance of nitric oxide (NO) in bioregulatory processes and other physiological functions prompted the development of photoprotecting groups specifically designed for its release. A most recent example is the synthesis and photochemistry of a series of naphthylmethyl and naphthylallyl diazeniumdiolates.76 These derivatives, represented by 79 in Eq. (69.40), were prepared from reaction of 1-(N,Ndiethylamino)-diazen-1-ium-1,3-diolate (81) with the corresponding alkyl bromide. Photolysis produced a mixture of products that resulted from two different reaction pathways. Path a is a nonproductive pathway that leads to the formation of nitrosamine 80 and oxime byproducts. Path b leads to diazeniumdiolate 81, which collapses to give free NO, along with diethylamine and other photoproducts. The extent to which the reaction follows one pathway over another is dependent on the substituents present on the naphthyl ring. As Table 69.22 shows, the best deprotection yields were obtained with a methoxy group at the 5 and 8 positions of the ring. 1348_C69.fm Page 35 Monday, October 13, 2003 3:22 PM 69-35 Photoremovable Protecting Groups TABLE 69.22 NO Cages and Their Corresponding Deprotection Yields and Quantum Efficiencies NO cage Protection (% Yield) Φ Deprotectiona,b (% Yield) 79a 79b 79c 79d 79e 90 25 30 36 5 0.007 Not determined 0.12 0.12 0.66 1 1 25 40 95 a b O X N O N NEt2 Based on the disappearance of starting material (using HPLC) and the amount of NO measured; thermal decomposition of 81 was found to produce 1.5 equivalents of NO. For 79a–c, a wavelength of 300 nm was used; for 79d,e, the wavelength was 350 nm. a O Et2N N n hν n N O oximes 80 CH3CN(aq) X Ar + Y 79 b Et2N O N N O + Ar 2 NO CH2 n + n = 0,1 81 + other photoproducts n = 0, X = Y = H n = 0, X = H, Y = Me n = 1, X = Y = H n = 0, X = OMe, Y = H n = 1, X = OMe, Y = H 79a 79b 79c 79d 79e Et2NH (69.40) The likely mechanism proceeds through photosolvolysis of the carbon–oxygen bond, resulting in a resonance-stabilized carbocation. Subsequent release of NO occurs from the diazeniumdiolate 81. Acidic conditions that protonated the amine greatly enhanced the rate of NO release. Derivative 79e undergoes clean deprotection and exhibits an excellent quantum efficiency, making it the most attractive of the NO cages studied thus far. Some additional advantages are its absorption beyond 300 nm (λmax = 336 nm) and its stability in acidic and basic solutions at room temperature for up to 24 hr. Unfortunately, it suffers from a low protection yield of only 5%, and its solubility is limited to 20 µM in 95% aqueous acetonitrile. Despite these shortcomings, its development may pave the way for similar methoxy-substituted naphthylallyl derivatives with increased solubility in aqueous media and higher protection yields. A unique photoprotecting group for alcohols and thiols was reported in the mid 1990s.77 Benzoylbenzoate ester 82 (Eq. (69.41)) was synthesized in one step from DCC coupling of the corresponding alcohol or thiol to 2-benzoylbenzoic acid. Photolysis at ~300 nm in the presence of cyclohexylamine, an electron donor, afforded 3-phenylphthalide 84 along with the free alcohol or thiol. O Ph CO2R OH hν cyclohexylamine CO2R O + Ph Ph O ROH or RSH 1:1 PhH:CH3CN 82 83 84 R = n-C12H25-, c-C12H23-, cholesteryl-, geranyl-, 2',3'-isopropylidene uridinyl-, n-C12H25 (thioester) (69.41) 1348_C69.fm Page 36 Monday, October 13, 2003 3:22 PM 69-36 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition *3 O Ph O hν Ar Ph ISC NH2 NH2 O Ar Ph Ar Ar = o-benzoate ester NH2 Ph OH OH Ar 85 NH NH2 OH Ph Ph Ar Ar + 83 86 SCHEME 12 Proposed photoreduction mechanism of benzoylbenzoate esters.78 TABLE 69.23 Data for Benzoylbenzoate Esters R n-C12H25c-C12H23CholesterylGeranyl2′,3′-isopropylidene uridinyl n-C12H25 (thioester) a b c Protection (% Yield) Deprotectiona (% Yield) 76 50 67 63 79 76 95 85 100 90b 90c 60 Yield of the recovered alcohol (thiol) was determined with NMR. sec-Butylamine was used as the electron donor; the solvent was 1:1 benzene/isopropanol. sec-Butylamine was the electron donor; photolysis was carried out with a uranium filter. The mechanism outlined in Scheme 1278 involves electron transfer from the amine to the ketone in the excited state followed by intermolecular proton transfer to generate radical pair 85–86; a second electron transfer and proton exchange lead to the reduced alcohol 83, which lactonizes to form 84 concurrently with release of the alcohol. In general, the benzoylbenzoate photoprotecting group worked well for the particular substrates studied. Synthesis yields for the benzoylbenzoate cages were respectable, and the deprotection occurred in most cases with good recoveries of the alcohol (Table 69.23). Problems were encountered in the photolysis of the caged thiol that led to the formation of side products and thus a lower overall deprotection yield. While the benzoylbenzoate cages exhibit good deprotection yields for alcohols, their application is limited to organic solvents. The presence of an electron donor (i.e., aliphatic amine) is also required, a necessity that may complicate the reaction mixture in the presence of other sensitive functional groups. Finally, the release process must be inherently a slow one due to the ground state lactonization process involved. 69.7 Phenols and Other Weak Acids o-Nitrobenzyl The photolabile o-nitrobenzyl derivative was utilized to protect the phenolic OH group of serotonin.79 The serotonin type-3 receptor is the only ligand-gated ion channel in the 5-HT receptor family.80,81 The protection of the phenolic hydroxy group of serotonin required four steps, as shown in Scheme 13. The substrate was released upon excitation with 337-nm laser pulses. The signal decay from pulsed laser 1348_C69.fm Page 37 Monday, October 13, 2003 3:22 PM 69-37 Photoremovable Protecting Groups NH2 HO NO2 COOCH3 O 1) di-t-BOC anhydride (97%) N H 2) K+ t-butylate N H N H NO2 COOCH3 Serotonin BOC N-Boc-OMeCNB-5HT Br 1) 3.5% K2CO3 MeOH/H2O 1:1 (85%) Methyl-2-bromo-2(2-nitrophenyl) acetate 2) FCH2COOH CH2Cl2/Acetone/H2O 10:10:1 (90%) (15%) NO NO2 COOH O COOH hν, 337 nm O + Serotonin NH2 -H+ N H O-CNB-5HT SCHEME 13 Protection and photochemical deprotection of serotonin. studies gave a time constant of 16 µs, and the quantum yield was determined to be 0.03. The rate of decay of the intermediate was observed to be pH dependent. The caged serotonin showed good solubility in buffered aqueous media (in excess of 2 mM); however, the authors suggested that the caged compound was subject to hydrolysis in the dark on standing. This photoremovable protecting group has been employed as a photocleavable linker to reagents bound to Au surfaces.82 4-Hydroxy-stilbene was linked to 6-bromohexyl-3-nitro-4-bromomethylbenzoate in 51% yield, then thiolated by trimethylsilylthioxy dehalogenation in THF, followed by desilylation in situ. The self-assembled monolayers of long-chain alkyl thiolate on bulk polycrystalline gold were constructed. Upon irradiation at 350 nm, the Z,E-photoisomerization attained a photostationary state within 25 min, while the dissociation took about 60 min; however, sensitization with 1,4-dibromonaphthalene (ET = 58.1 kcal/mol) produced a cleaner photoisomerization. The unidirectional isomerization, from cis to trans, by both direct irradiation and sensitization was followed by the release of a bound chain from the metal surface. O O hν, 350 nm ON HO + O2N O O O(CH2)6SH O(CH2)6SH hν, 350 nm hν, 350 nm O O HO hν, 350 nm O2N ON + O O O(CH2)6SH SCHEME 14 O(CH2)6SH 1348_C69.fm Page 38 Monday, October 13, 2003 3:22 PM 69-38 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition Benzoin The ubiquinol oxidizing enzyme is a redox active enzyme that requires a fast two-electron reduction of ubiquinone. 3′,5′-Dimethoxybenzoin (DMB) caged ubiquinol83 was synthesized to study the detailed enzymatic mechanism of the fast electron-transfer process in redox active enzymes. The monosilylated ubiquinol was coupled to the protecting group to form the o-nitrophenylcarbonate ester of 3′,5′dimethoxyphenyl(phenyldithiane) in 55% yield. Upon photolysis, DMB caged ubiquinol generated ubiquinone with a rate greater than 106 s–1(Eq. (69.42)). O OH H3CO H3CO CH3 CH3 H3CO H3CO O O hn, 355 nm O O -CO2 OCH3 + H3CO O O OCH3 OCH3 11 (69.42) Pirrung and Bradley70 also applied DMB (3′,5′-dimethoxybenzoin) to protect the phenolic group, demonstrating that 4-methoxyphenol was released upon irradiation at 350 nm in 90% yield (Eq. (69.43)). H3CO O C O O hn, 350 nm H3CO H3CO H3CO Ph + T HF O OH OCH3 -CO2 11 90% O OCH3 (69.43) 69.8 Amines o-Nitrobenzyl There are few variations for effective amine photoremovable protecting groups. The o-nitrobenzyl group remains the most popular group and among the many examples the studies by Cameron and Frechet are noteworthy.84 In general, o-nitrobenzylcarbamates of aliphatic amines upon photolysis release the amine in good yield. For example, cyclohexylamine is released from its o-nitrobenzyl carbamate 87 as the corresponding free carbamate upon irradiation in THF at 254 nm (Eq. (69.44)). NO2 R2 O O hν, 254 nm N H NO R2 O THF + CO2 + H2N R1 R1 87 (69.44) Subsequent loss of carbon dioxide frees the amine. Quantum efficiencies varied depending on the substituents present at the ortho and benzylic positions (Table 69.24). The best efficiencies were obtained with two o-nitro groups on the aryl ring, likely increasing the probability for the hydrogen atom abstraction by one of the nitro groups. 1348_C69.fm Page 39 Monday, October 13, 2003 3:22 PM 69-39 Photoremovable Protecting Groups TABLE 69.24 Protection Yieldsa and Quantum Efficiencies for Various Substituted o-Nitrobenzyl Carbamates R1 R2 H NO2 H NO2 H NO2 H H Me Me o-Nitrophenyl 2,6-Dinitrophenyl a Protection (% Yield) Φ 71 78 71 71 79 52 0.13 0.62 0.11 0.35 0.26 0.28 Deprotection yields were not provided. A problem that is not entirely avoidable is the formation of imine byproducts via reaction of the released amine with the aldehydic group in the photoproduct. This occurrence could be suppressed with alkyl or aryl substitution at the benzylic position, leading to the formation of a less reactive ketone in comparison with the nitroso aldehyde formed with no substitution at the benzylic position. Imine byproduct formation is also less likely to occur in relatively nonpolar solvent systems, such as THF, which ultimately limits the application of the o-nitrobenzyl carbamate photoprotecting group to nonaqueous systems in this regard. Benzoin Derivatives In the mid-1990s, Pirrung and Huang85 extended the use of the benzoin photoprotecting group to the release of amines by synthesizing m,m′-dimethoxybenzoin (DMB) carbamates. The DMB derivatives were synthesized by coupling the corresponding amine with benzoin carbonyl chloride that had been elaborated by reaction of carbonyldiimidazole with the methyl triflate of 88 followed by nucleophilic addition of DMB. Irradiation in benzene or THF at 350 nm produced the free amine, carbon dioxide, and the benzofuran byproduct (Eq. (69.45)). R2 N O R1 hν, 350 nm O R1NHR2 MeO benzene + 11 + CO2 O OMe 88 (69.45) Five different amines were protected and recovered in moderate to good yields as shown in Table 69.25. The DMB carbamates appeared to work well for a variety of amines in the presence of other functional groups, such as alcohols or esters; however, the reaction is limited to secondary amines, as primary amines were found to undergo intramolecular cyclization leading to byproducts that are inert to photolysis. Arylsulfonamides Corrie and Papageorgiou86 have reported the synthesis and photochemistry of various methoxysubstituted arylsulfonamides. Similar derivatives had been previously found to undergo single electron transfer reactions in the excited state, leading to cleavage of the sulfur–nitrogen bond.87,88 It was reasoned that such a process could be used for the rapid release of neurologically active amines. The arylsulfonamide derivatives were synthesized in several steps, starting from 1,5-dimethoxynaphthalene. Photolysis of 89 in phosphate buffer (pH 7.0) in the presence of ascorbate resulted in release of 1348_C69.fm Page 40 Monday, October 13, 2003 3:22 PM 69-40 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition TABLE 69.25 Protection and Deprotection Yields for DMB Carbamates Amine Protection (% Yield) Deprotection (% Yield) 85 89a 76 79 N H 90 56 CO2t-Bu 90 73 88 97a NH N H N H HO Ph N H a The amine was recovered as the corresponding hydrochloride salt. TABLE 69.26 Synthesis and Photochemistry Yields of Arylsulfonamides R1 (CH2)3OPO3–2 (CH2)3OPO3–2 Me a b R2 Protection (% Yield) Deprotectiona (% Yield) 71 61 59 35 22b 53 CO2– CH2CO2– CO2Me Yields were determined using quantitative amino acid analysis; irradiations were only carried out to approximately 50% conversion of the starting material in the presence of 10-mM ascorbate. Photolyzed in buffer solution only. the free amine in low to moderate yields, accompanied by the reduced arylsulfonyl byproduct 90 (Eq. (69.46) and Table 69.26). OMe OMe hν 1:2 buffer/MeOH ascorbate OR1 SO2NH 89 + H2N R2 OR1 SO2- R2 90 (69.46) 1348_C69.fm Page 41 Monday, October 13, 2003 3:22 PM 69-41 Photoremovable Protecting Groups OMe OMe 89 hν H2O SET -R2CH2NH2 back electron transfer 90 OR1 OR1 O S O O NHCH2R2 S O SCHEME 15 Proposed mechanism for amino acid release from arylsulfonamides. The mechanism is thought to involve an excited state intramolecular single eletron trnasfer from the electron-rich naphthalene to the sulfonamide group (Scheme 15); release of the amine then occurs via assistance from the neighboring oxygen, leading to a radical cation that is subsequently reduced to 90 by ascorbate. It was speculated that, in the case of the glycine and β-alanine substrates, a competitive electron transfer was occurring from the carboxylate group to the radical cation of 90. Such a process would yield a carboxyl radical that would undergo subsequent decarboxylation, leading to a mixture of side products and thus a lower yield of the free amino acid. Spectroscopic evidence using LFP combined with FTIR spectroscopy supported this hypothesis. Low yields were still encountered even in the presence of increased amounts of ascorbate, suggesting that the electron transfer was taking place within a solvent cage. Despite this shortcoming, the arylsulfonamide group may still hold promise as a photoremovable protecting group for amines lacking a carboxylate moiety. Further studies would need to be carried out to fully establish its capabilities in this regard. 69.9 Conclusion A wide variety of photoremovable protecting groups have been added to the veteran o-nitrobenzyl series. Each new group has been developed to address the shortcomings of the o-nitrobenzyl group or to add features such as faster rates for release, extension of the absorption range into the near UV-visible region, improved solubility, higher efficiencies, improved conversions and yields, and more benign photoproducts from the protecting group. Extensions and applications of this chemistry to two photon excitation processes, to traceless reagents in combinatorial chemistry and photolithography, as orthogonal reagents in synthesis, and to time-resolved spectroscopic techniques wil make even more demands on the design, synthesis, and development of new photoremovable protecting groups. Even now, however, no single photoremovable protecting group fulfills all nine criteria Sheehan and Lester had suggested (outlined at the beginning of this chapter). Nevertheless, important progress has been achieved as evidenced by the growing number of applications reported for many of these photoremovable groups, especially in biological studies. Applications in synthesis, combinatorial chemistry, micro arrays, and photolithography are also forthcoming. These fields have benefited and will continue to draw the interest of the science community as improvements of existing systems and discovery of new photoactive protecting groups are developed. This field of photoremovable protecting groups is still in its infancy. Acknowledgments We acknowledge the support of the Department of Energy, University of Kansas and the National Science Foundation. 1348_C69.fm Page 42 Monday, October 13, 2003 3:22 PM 69-42 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition References and Notes 1. (a) Kaplan, J. H., Forbush, B. I., and Hoffman, J. F., Rapid photolytic release of adenosine 5′triphosphate from a protected analogue: utilization by the Na:K pump of human red blood cell ghosts, Biochemistry, 17, 1929–1935, 1978. (b) Engels, J. and Schlaeger, E.-J., Synthesis, structure, and reactivity of adenosine cyclic 3′,5′-phosphate benzyl triesters, J. Med. Chem., 20, 907-911, 1977. 2. Givens, R. S. and Kueper III, L. W., Photochemistry of phosphate esters, Chem. Rev., 93, 55–66, 1993. 3. Adams, S. R. and Tsien, R. Y., Controlling cell chemistry with caged compounds, Ann. Rev. Physiol., 18, 755–784, 2000. 4. Corrie, J. E. T. and Trentham, D. R., Synthetic, mechanistic and photochemical studies of phosphate esters of substituted benzoins, J. Chem. Soc., Perkin Trans. 1, 2409–2417, 1992. 5. Pelliccioli, A. P. and Wirz, J., Photoremovable-protecting groups: reaction mechanism and applications, Photochem. Photobiol. Sci., 1, 441–458, 2002. 6. Bochet, C. G., Photolabile protecting groups and linkers, J. Chem. Soc., Perkin Trans. 1, 125–142, 2002. 7. Givens, R. S. and Lee, J.-I., The p-hydroxyphenacyl photoremovable protecting group, J. Photosci., 2002, in press. 8. Zou, K., Miller, W. T., Givens, R. S., and Bayley, H., Caged thiophosphotryosine peptides, Angew. Chem. Int. Ed. Engl., 40, 3049–3051, 2001. 9. Zou, K., Cheley, S., Givens, R. S., and Bayley, H., Catalytic subunit of protein kinase caged at the activating phosphothreonine, J. Am. Chem. Soc., 124, 8220–8229, 2002. 10. Arabaci, G., Guo, X.-C., Beebe, K. D., Coggeshall, K. M., and Pei, D., α-Haloacetophenone derivatives as photoreversible covalent inhibitors of protein tyrosine phosphates, J. Am. Chem. Soc., 121, 5085–5086, 1999. 11. Corey, E. J. and Cheng, X.-M., The Logic of Chemical Synthesis, John Wiley & Sons, New York, 1995, 78–79. 12. Sheehan, J. C. and Umezawa, K., Phenacyl photosensitive blocking groups, J. Org. Chem., 38, 3771–3774, 1973. 13. Barltrop, J. A. and Schofield, P., Photosensitive protecting groups, Tetrahedron Lett., 697–699, 1962. 14. Givens, R. S., Weber, J. F. W., Jung, A. H., and Park, C.-H., New photoprotecting groups: desyl and p-hydroxyphenacyl phosphate and carboxylate esters, in Methods in Enzymology, Vol. 291, Marriott, G., Ed., Academic Press, New York, 1998, 1–29. 15. Baldwin, J. E., McConnaughie, A. W., Moloney, M. G., Pratt, A. J., and Shim, S. B., New photolabile phosphate protecting groups, Tetrahedron, 46, 6879–6884, 1990. 16. Wang, B. and Zheng, A., A photosensitive protecting group for amines based on coumarin chemistry, Chem. Pharm. Bull., 45, 715–718, 1997. 17. Koenigs, P. M., Faust, B. C., and Porter, N. A., Photochemistry of enzyme-bound cinnamoyl derivatives, J. Am. Chem. Soc., 115, 9371–9379, 1993. 18. Lester, H. A. and Nerbonne, J. M., Physiological and pharmacological manipulations with light flashes, Ann. Rev. Biophys. Bioeng., 11, 151–175, 1982. 19. Barltrop, J. A., Plant, P. J., and Schofield, P., Photosensitive protective groups, J. Chem. Soc., Chem. Commun., 822–823, 1966. 20. Ried, V. W. and Wilk, M., The photochemistry of unsaturated nitrocompounds. II. Relations among polarizability, reduction potential and photochemical behavior of some nitro compounds, Justus Liebigs Ann. Chem., 590, 91–110, 1954. 21. Walker, J. W., Lu, Z., and Moss, R. L., Effects of Ca+2 on the kinetics of phosphate release in skeletal muscle, J. Biol. Chem., 267, 2459–2466, 1992. 22. Willenbucher, R. F., Xie, Y. N., Eysselein, V. E., and Snape, Jr., W. J., Mechanisms of cAMP-mediated relaxation of distal circular muscle in rabbit colon, Am. J. Physiol., 262, G159–G164, 1992. 23. Ostap, E. M. and Thomas, D. D., Rotational dynamics of spin-labeled F-actin during activation of myosin S1 ATPase using caged ATP, Biophys. J., 59, 1235–1241, 1991. 1348_C69.fm Page 43 Monday, October 13, 2003 3:22 PM Photoremovable Protecting Groups 69-43 24. Tepe, J. J. and Williams, R. M., DNA crosslinking by a phototriggered dehydromonocrotaline progenitor., J. Am. Chem. Soc., 121, 2951–2955, 1999. 25. Sheehan, J. C. and Wilson, R. M., Photolysis of desyl compounds: a new photolytic cyclization, J. Am. Chem. Soc., 86, 5277–5281, 1964. 26. Givens, R. S. and Matuszewski, B., Photochemistry of phosphate esters: an efficient method for the generation of electrophiles, J. Am. Chem. Soc., 106, 6860–6861, 1984. 27. Givens, R. S., Athey, P. S., Kueper, L. W. I., Matuszewski, B., and Xue, J.-Y., Photochemistry of αketo phosphate esters: photorelease of a caged cAMP, J. Am. Chem. Soc., 114, 8708–8710, 1992. 28. Givens, R. S., Athey, P. S., Matuszewski, B., Kueper, L. W., Xue, J., and Fisher, T., Photochemistry of phosphate esters: α-keto phosphates as a photoprotecting group for caged phosphate, J. Am. Chem. Soc., 115, 6001–6012, 1993. 29. Sheehan, J. C., Wilson, R. M., and Oxford, A. W., The photolysis of methoxy-substituted benzoin esters, J. Am. Chem. Soc., 93, 7222–7228, 1971. 30. Rajesh, C. S., Givens, R. S., and Wirz, J., Kinetics and mechanism of phosphate photorelease from benzoin diethyl phosphate: evidence for adiabatic fission to an α-keto cation in the triplet state, J. Am. Chem. Soc., 122, 611–618, 2000. 31. Epstein, W. W. and Garrossian, M., p-Methoxyphenacyl esters as photodeblockable-protecting groups for phosphates, J. Chem. Soc., Chem. Commun., 532–533, 1987. 32. Dhavale, D. D., Mali, V. P., Sudrik, S. G., and Sonawane, H. R., Media controlled photo-Favorskii type rearrangement of α-chloro acetophenones: synthesis of phenylacetic acids, Tetrahedron, 53, 16789–16794, 1997. 33. Sonawane, H. R., Bellur, N. S., Kulkarni, D. G., and Ayyangar, N. R., Photochemical rearrangement of α-chloropropiophenones to α-arylpropanoic acids: studies on chirality transfer and synthesis of (S)-(+)-ibuprofen and (S)-(+)-ketoprofen, Tetrahedron, 50, 1243–1260, 1994. 34. Anderson, J. C. and Reese, C. B., A photo-induced rearrangement involving aryl participation, Tetrahedron Lett., 1–4, 1962. 35. Givens, R. S. and Park, C.-H., p-Hydroxyphenacyl ATP: a new phototrigger, Tetrahedron Lett., 37, 6259–6262, 1996. 36. Park, C.-H. and Givens, R. S., New photoactivated protecting groups. 6. p-Hydroxyphenacyl: a phototrigger for chemical and biochemical probes J. Am. Chem. Soc., 119, 2453–2463, 1997. 37. Zhang, K., Corrie, J. E. T., Munasinghe, R. N., and Wan, P., Mechanism of photosolvolytic rearrangement of p-hydroxyphenacyl esters: evidence for excited-state intramolecular proton transfer as the primary photochemical step, J. Am. Chem. Soc., 121, 5625–5632, 1999. 38. (a) Fischer, M. and Wan, P., m-Quinone methides from m-hydroxy-1,1-diaryl alkenes via excitedstate (formal)intramolecular proton transfer mediated by a witer timer, J. Am. Chem. Soc., 120, 2680-2681, 1998. (b) Kalanderopoulos, P. and Yates, P., Intramolecular proton transfer in photohydration reactions, J. Am. Chem. Soc., 108, 6290-6295, 1986. 39. Conrad II, P. G., Givens, R. S., Hellrung, B., Rajesh, C. S., Ramseiir, M., and Wirz, J., p-Hydroxyphenacyl phototriggers: the reactive excited state of phosphate photorelease, J. Am. Chem. Soc., 122, 9346–9347, 2000. 40. Zimmerman, H. E. and Sandel, V. R., Mechanistic organic photochemistry. II. Solvolytic photochemical reactions, J. Am. Chem. Soc., 85, 915–922, 1963. 41. Zimmerman, H. E., Meta-ortho effect in organic photochemistry: mechanistic and exploratory organic photochemistry, J. Phys. Chem. A, 102, 5616-5621, 1998. (See also, Zimmerman, H. E., Meta-ortho effect in organic photochemistry: mechanistic and exploratory organic photochemistry. [Erratum to document cited in CA129:81353] J. Phys. Chem. A, 102, 7725, 1998.) 42. DeCosta, D. P. and Pincock, J. A., Photochemistry of substituted 1-naphthylmethyl esters of phenylacetic and 3-phenylpropanoic acid: radical pairs, ion pairs and Marcus electron transfer, J. Am. Chem. Soc., 115, 2180–2190, 1993. 43. Pincock, J. A., Photochemistry of arylmethyl esters in nucleophilic solvents: radical pair and ion pair intermediates, Acc. Chem. Res., 30, 43–49, 1997. 1348_C69.fm Page 44 Monday, October 13, 2003 3:22 PM 69-44 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition 44. Furuta, T., Momotaka, A., Sugimoto, M., Hatayajma, M., Torigai, H., and Iwamura, M., Acyloxycoumarinylmethyl-caged cAMP: the photolabile and membrane-permeable derivative of cAMP that effectively stimulates pigment-dispersion response of melanophores, Biochem. Biophys. Res. Commun., 228, 193–198, 1996. 45. Gee, K. R., Niu, L., Schaper, K., Jayaraman, V., and Hess, G. P., Synthesis and photochemistry of a photolabile precursor of N-methyl-D-aspartate (NMDA) that is photolyzed in the microsecond time region and is suitable for chemical kinetic investigations of the NMDA receptor, Biochemistry, 38, 3140–3147, 1999. 46. Yoo, D. J. and Greenberg, M., Synthesis of oligonucleotides containing 3′-alkyl carboxylic acids using universal, photolabile solid phase synthesis supports, J. Org. Chem., 60, 3358–3364, 1995. 47. Schade, B., Hagen, V., Schmidt, R., Herbrich, R., Krause, E., Eckardt, T., and Bendig, J., Deactivation behavior and excited-state properties of (coumarin-4-yl)methyl derivatives. 1. Photocleavage of (7-methoxycoumarin-4-yl)methyl caged acids with fluorescence enhancement, J. Org. Chem., 64, 9109–9117, 1999. 48. Conrad II, P. G., Givens, R. S., Weber, J. F. W., and Kandler, K., New phototriggers: extending the p-hydroxyphenacyl π-π* absorption range, Org. Lett., 2, 1545–1547, 2000. 49. Givens, R. S., Weber, J. F. W., Conrad II, P. G., Orosz, G., Donahue, S. L., and Thayer, S. A., New phototriggers. 9. p-Hydroxyphenacyl as a C-terminal photoremovable protecting group for oligopeptides, J. Am. Chem. Soc., 122, 2687–2697, 2000. 50. Routledge, A., Abell, C., and Balasubramanian, S., The use of a dithiane protected benzoin photolabile safety catch linker for solid-phase synthesis, Tetrahedron Lett., 38, 1227–1230, 1997. 51. Klan, P., Zabadal, M., and Heger, D., 2,5-Dimethylphenacyl as a new photoreleasable-protecting group for carboxylic acids, Org. Lett., 2, 1569–1571, 2000. 52. Zabadal, M., Pelliccioli, A. P., Klan, P., and Wirz, J., 2,5-Dimethylphenacyl esters: a photoremovable protecting group for carboxylic acids, J. Phys. Chem. A, 105, 10329–10333, 2001. 53. Bochet, C. G., Orthogonal photolysis of protecting groups, Angew. Chem., Int. Ed. Engl., 40, 2071–2073, 2001. 54. Walker, J. W., Reid, G. P., McCray, J. A., and Trentham, D. R., Photolabile 1-(2-nitrophenyl)ethyl phosphate esters of adenine nucleotide analogues: synthesis and mechanism of photolysis, J. Am. Chem. Soc., 110, 7170–7177, 1988. 55. Furuta, T., Torigai, M., Sugimoto, M., and Iwamura, M., Photochemical properties of new photolabile cAMP derivatives in a physiological saline solution, J. Org. Chem., 60, 3953–3956, 1995. 56. Furuta, T. and Iwamura, M., New caged groups: 7-substituted coumarinylmethyl phosphate esters, in Methods in Enzymology, Vol. 291, Marriott, G., Ed., Academic Press, New York, 1998, 50–63. 57. Hagen, V., Bendig, J., Frings, S., Eckardt, T., Helm, S., Reuter, D., and Kaupp, U. B., Highly efficient and ultrafast phototriggers for cAMP and cGMP by using long-wavelength UV/VIS activation, Angew. Chem., Int. Ed. Engl., 40, 1046–1048, 2001. 58. Du, X., Frei, H., and Kim, S.-H., The mechanism of GTP hydrolysis by Ras probed by Fourier transform infrared spectroscopy, J. Biol. Chem., 275, 8492–8500, 2000. 59. Geibel, S., Barth, A., Amslinger, S., Jung, A. H., Burzik, C., Clarke, R. J., Givens, R. S., and Fendler, K., P3-[2-(4-hydroxyphenyl)-2-oxo]ethyl ATP for the rapid activation of the Na+, K+-ATPase, Biophys. J., 79, 1346–1357, 2000. 60. Pirrung, M. C. and Shuey, S. W., Photoremovable-protecting groups for phosphorylation of chiral alcohols: asymmetric synthesis of phosphotriesters of (-)-3′,5′-dimethoxybenzoin, J. Org. Chem., 59, 3890–3897, 1994. 61. Watanabe, S., Sueyoshi, T., Ichihara, M., Uehara, C., and Iwamura, M., Reductive ring opening of o-nitrobenzylidene acetals of monosaccharides: synthesis and photolysis of some photolabile sugars, Org. Lett., 3, 255–257, 2001. 62. Smith, A. B., Savinov, S. N., Manjappara, U. V., and Chaiken, I. M., Peptide–small molecule hybrids via orthogonal deprotection-chemoselective conjugation to cysteine-anchored scaffolds: a model study, Org. Lett., 4, 4041–4044, 2002. 1348_C69.fm Page 45 Monday, October 13, 2003 3:22 PM Photoremovable Protecting Groups 69-45 63. Hasan, A., Stengele, K.-P., Giegrich, H., Cornwell, P., Isham, K. R., Sachleben, R. A., Pfleiderer, W., and Foote, R. S., Photolabile protecting groups for nucleosides: synthesis and photodeprotection rates, Tetrahedron, 53, 4247–4264, 1997. 64. Giegrich, H., Eisele-Buhler, S., Hermann, C., Kvasyuk, E., Charubala, R., and Pfleiderer, W., New photolabile protecting groups in nucleoside and nucleotide chemistry: synthesis, cleavage mechanisms and applications, Nucleosides & Nucleotides, 17, 1987–1996, 1998. 65. Walbert, S., Pfleiderer, W., and Steiner, U. E., Photolabile protecting groups for nucleosides: mechanistic studies of the 2-(2-nitrophenyl)ethyl group, Helv. Chim. Acta, 84, 1601–1611, 2001. 66. Pirrung, M. C., Wang, L., and Montague-Smith, M. P., 3′-Nitrophenylpropyloxycarbonyl (NPPOC) protecting groups for high-fidelity automated 5′ → 3′ photochemical DNA synthesis, Org. Lett., 3, 1105–1108, 2001. 67. Coleman, M. P. and Boyd, M. K., The S-pixyl group: an efficient photocleavable-protecting group for the 5′ hydroxy function of deoxyribonucleosides, Tetrahedron Lett., 40, 7911–7915, 1999. 68. Lin, W. and Lawrence, D. S., A strategy for the construction of caged diols using a photolabile protecting group, J. Org. Chem., 67, 2723–2726, 2002. 69. Intermolecular ion pairs have been observed in the photolysis of caged methoxycoumarin derivatives; see Reference 47. 70. Pirrung, M. C. and Bradley, J.-C., Dimethoxybenzoin carbonates: photochemically removable alcohol protecting groups suitable for phosphoramidite-based DNA synthesis, J. Org. Chem., 60, 1116–1117, 1995. 71. Furuta, T., Hirayama, Y., and Iwamura, M., Anthraquinon-2-ylmethoxycarbonyl (Aqmoc): a new photochemically removable-protecting group for alcohols, Org. Lett., 3, 1809–1812, 2001. 72. Loudwig, S. and Goeldner, M., N-methyl-N-(o-nitrophenyl)carbamates as photolabile alcohol protecting groups, Tetrahedron Lett., 42, 7957–7959, 2001. 73. The carbamate cage is inert to strong non-nucleophilic bases such as LDA. 74. Pirrung, M. C., Fallon, L., Zhu, J., and Lee, Y. R. Photochemically removable silyl protecting groups, J. Am. Chem. Soc., 123, 3638–3643, 2001. 75. Pirrung, M. C. and Lee, Y. R., Photochemically removable silyl protecting groups, J. Org. Chem., 58, 6961–6963, 1993. 76. Bushan, K. M., Xu, H., Ruane, P. H., D’Sa, R. A., Pavlos, C. M., Smith, J. A., Celius, T. C., and Toscano, J. P., Controlled photochemical release of nitric oxide from O2-naphthylmethyl- and O2naphthylallyl-substituted diazeniumdiolates, J. Am. Chem. Soc., 124, 12640–12641, 2002. 77. Jones, P. B., Pollastri, M. P., and Porter, N. A., 2-Benzoylbenzoic acid: a photolabile mask for alcohols and thiols, J. Org. Chem., 61, 9455–9461, 1996. 78. Based on the photoreduction mechanism proposed for benzophenone in the presence of amines, Cohen, S. G. and Chao, H. M., Photoreduction of aromatic ketones by amines: studies of quantum yields and mechanism, J. Am. Chem. Soc., 90, 165–173, 1968. 79. Breitinger, H. A., Wieboldt, R., Ramesh, D., Carpenter, B., and Hess, G., Synthesis and characterization of photolabile derivatives of serotonin for chemical kinetic investigations of the serotonin 5-HT3 receptor, Biochemistry, 39, 5500–5508, 2000. 80. Derkach, V., Surprenant, A., and North, R. A., 5-HT3 receptors are membrane ion channels, Nature, 339, 706–709, 1989. 81. Tyers, M. B., 5-HT3 receptors, Ann. N.Y. Acad. Sci., 600, 194–205, 1990. 82. Hu, J., Zhang, J., Liu, F., Kittredge, K., Whitesell, J. K., and Fox, M. A., Competitive photochemical reactivity in a self-assembled monolayer on a colloidal gold cluster, J. Am. Chem. Soc., 123, 1464–1470, 2001. 83. Stowell, M. H. B., Wang, G., Day, M. W., and Chan, S. I., Design, synthesis and photochemical properties of a photoreleasable ubiquinol-2: a novel compound for studying rapid electron-transfer kinetics in ubiquinol-oxidizing enzymes, J. Am. Chem. Soc., 120, 1657–1664, 1998. 84. Cameron, J. F. and Frechet, J. M. J., Photogeneration of organic bases from o-nitrobenzyl derived carbamates, J. Am. Chem. Soc., 113, 4303–4313, 1991. 1348_C69.fm Page 46 Monday, October 13, 2003 3:22 PM 69-46 CRC Handbook of Organic Photochemistry and Photobiology, 2nd Edition 85. Pirrung, M. C. and Huang, C.-Y., Photochemical deprotection of 3′,5′-dimethoxybenzoin (DMB) carbamates derived from secondary amines, Tetrahedron Lett., 36, 5883–5884, 1995. 86. Corrie, J. E. T. and Papageorgiou, G., Synthesis and evaluation of photolabile sulfonamides as potential reagents for rapid photorelease of neuroactive amines, J. Chem. Soc., Perkin Trans. 1, 1583–1591, 1996. 87. Hamada, T., Nishida, A., and Yonemitsu, O., Selective removal of electron-accepting p-toluene and naphthalenesulfonyl protecting groups for amino function via photoinduced donor–acceptor ion pairs with electron donating aromatics, J. Am. Chem. Soc., 108, 140–145, 1986. 88. Hamada, T., Nishida, A., and Yonemitsu, O., A new amino protecting group readily removable with near ultraviolet light as an application of electron-transfer photochemistry, Tetrahedron Lett., 30, 4241–4244, 1989.