Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Point mutation wikipedia , lookup
Evolution of metal ions in biological systems wikipedia , lookup
Genetic code wikipedia , lookup
Fatty acid metabolism wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Oligonucleotide synthesis wikipedia , lookup
Oxidative phosphorylation wikipedia , lookup
Specialized pro-resolving mediators wikipedia , lookup
Peptide synthesis wikipedia , lookup
Fatty acid synthesis wikipedia , lookup
Butyric acid wikipedia , lookup
Adenosine triphosphate wikipedia , lookup
Nucleic acid analogue wikipedia , lookup
Biochemistry wikipedia , lookup
Citric acid cycle wikipedia , lookup
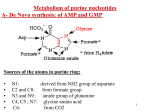
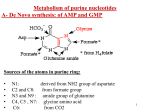
METABOLISM OF PURINE NUCLEOTIDES & PURINE DISORDERS ROLES OF NUCLEOTIDES Building blocks for DNA and RNA “Second messengers” in signal transduction cascades Energy “currency” of the cell Components of major co-enzymes Purine Biosynthesis • Major site – (Adenine & Guanine) in the Liver. • Synthesis starting from Ribose-5-phosphate Denova pathway: Precursors contribute to form the Purine ring: Glycine (C4, C5, N7), Glutamine ( N3, N9), THF (C2, C8 atoms) Aspartate (N1 atom), & CO2 (C6 atom). • The construction of 6-membered ring forms Inosine- 5monophosphate (IMP) – 1st Purine. • Energetics – expenditure of 6 high energy bonds. Salvage pathway - Free & dietary purine can be converted into corresponding nucleotides (no need ATP). • RBC, Neutrophils & Brain cannot produce Purines by de novo synthesis. But by only salvage pathway (HGPRTase). Glucose-6-P pentose phosphate pathway ATP Ribose-5-phosphate AMP 5-phosphoribosylpyrophosphate (PRPP) PRPP synthetase Gln PRPP Amino acids: amidotransferase Glu Gly + Gln + Asp Inosine mono phosphate (IMP) 5-phospho-ribosylamine Cofactors: N10-formyl THF Figure 1. Synthesis of inosine monophosphate (IMP) NAD+ aspartate + GTP IMP GDP + Pi NADH Gln + ATP fumarate Glu+AMP+PPi guanosine monophosphate (GMP) Figure 2. adenosine monophosphate (AMP) GDP ADP GTP ATP Formation of AMP and GMP from IMP Ribose-5-phosphate PRPP synthetase PRPP PRPP amidotransferase 5-phospho-ribosylamine IMP allosteric inhibition GMP AMP GDP ADP Figure 3. Allosteric inhibition of purine biosynthesis; also ATP stimulates formation of AMP. Regulatory enzyme is PRPP amido transferase, controlled by feed back inhibition of nucleotides - IMP, AMP & GMP. GMP, GDP or GTP AMP, ADP or ATP Adenosine Guanosine NH3 adenosine deaminase Inosine Ribose-1-P purine nucleoside phosphorylase +PRPP Guanine HGPRT Ribose-1-P Hypoxanthine Xanthine xanthine oxidase +PRPP HGPRT xanthine oxidase GMP Figure 4. Degradation IMP Uric acid of purines to uric acid and salvage of purine bases via hypoxanthine-guanine phosphoribosyl transferase METABOLISM OF PURINE Glucose-6-P pentose phosphate pathway ATP Ribose-5-phosphate AMP X1a 5-phosphoribosylpyrophosphate (PRPP) PRPP synthetase Gln PRPP Amino acids: amidotransferase Glu Gly + Gln + Asp Inosine mono phosphate (IMP) 5-phospho--ribosylamine Cofactors: N10-formyl THF Figure 1. Hyperuricemia: Gout: X1a = PRPP synthetase defects associated with a superactive enzyme characterized by an increased Vmax or an enzyme with a reduced Km for ribose-5-P. Disorder of Purine catabolism • In humans, Purine rings are degraded to the metabolically inert uric acid. • Normal level of uric acid in blood is 2-7 mg/dl (Female) & 3-8 mg/dl (Male). • Uric acid excretion in urine is about 500 – 700 mg/day. 1. GOUT (Hyper uricemia) • An inherited metabolic disease • Incidence - 3 /1000 person • Defect - partial deficiency of HGPRTase or increased levels of PRPP caused by a hyperactive synthetase. • Causes - ↓ Synthesis of GMP and IMP & ↑ in PRPP levels --> ↑d Purine biosynthesis by de novo pathway. Gout 1. Primary gout: • Inborn error of metabolism due to over production of uric acid • Causes: Varient PRPP synthetase, PRPP gltamyl amido transferase, HGPRT deficiency, Glu-6-Pase defiecincy. 2. Secondary gout: • Various disease causing ↑or ↓uric acid excretion. • E.g.: Cancer, Psoriasis, Trauma, Starvation, Impairment in renal function leads to gout. GOUT Causes • Uricosuria • Accumulation of sodium urate crystals in the soft tissues called Tophi -causes painful Gouty - Arthritis. • Deposition of Na-urate crystals -Renal calculi • Clinical finding- Red faced, Acidosis & Renal damage. Treatment • Reduced dietary Purine intake & restrict alcohol is advised. • Uricosuric drugs. For example – Probenecid (Benemide), Salicylates and Halofenate. • Allopurinol (analog of hypoxanthine) competitively inhibits Xanthine oxidase & thus uric acid synthesis (Suicide inhibition) • Coichicine is an anti-inflammatory drug to relieve pain. Other useful drugs include Indomethacin, Naproxen, Brufen, Corticosteroids etc. 2. LESCH-NYHAN SYNDROME • • • • • X-linked recessive disorder of Purine metabolism. Incidence is 1 in 10,000 males Defect - Total lack of HGPRTase, salvage pathway Thus ↑ PRPP (de novo pathway), ↑ production of uric acid. Diagnosis - Murexide test (reddish deposit) to detect urine uric acid Clinical manifestations • ↑ Production of uric acid causes severe gout, poor growth, and renal failure due to Nephrolithiasis. • Neurological abnormalities such as Mental retardation, Aggressive behavior, learning disability & compulsive selfdestructive behavior. • Irresistible urge to bite their finger & lips (Self mutilation) • It shows that abnormal behavior can be caused by absence of a single enzyme. GMP, GDP or GTP AMP, ADP or ATP Adenosine Guanosine NH3 Inosine Purine nucleoside phosphorylase Ribose-1-P +PRPP Guanine Xanthine GMP Ribose-1-P Hypoxanthine HGPRTase X2 Adenosine deaminase Inhibited by allopurinol xanthine oxidase xanthine oxidase +PRPP HGPRTase X2 IMP Uric acid Figure 4. X2 = moderate defect (>50% activity) leading to gout; severe defect (very low activity) leads to Lesch-Nyhan syndrome. 3. Immunodeficiency diseases • Immunodeficiency diseases are associated with Purine degradation disorders. • Defect: Adenosine deaminase & Purine nucleoside phosphorylase involved in uric acid synthesis. • Causes: Severe combined immunodeficiency (SCID) involving T-cell & usually B-cell dysfunctions and thus impaired the immunity. • Uric acid synthesis ↓& tissue level of N.S & N.T↑ • Treatment: Transferring ADA gene by Gene therapy. 4. Hypo uricemia Hypoxanthine • When serum uric acid level is less than 2 mg/dl represents hypouricemia. • A rare congenital Xanthine oxidase deficiency • Incidence is 1 in 45,000. xanthine oxidase Xanthine xanthine oxidase Uric acid • It leads to the increased excretion of Xanthine & Hypoxanthine. • Causes: Excretion of xanthine in urine - Xanthinuria. Frequently causes the formation of Xanthine stones in the urinary tract. SYNTHESIS OF PYRIMIDINE NUCLEOTIDES & PYRIMIDINE DISORDERS SYNTHESIS OF PYRIMIDINE NUCLEOTIDES • Synthesis of the Pyrimidine is less complex than that of the purines, since the base is much simpler (6 member ring). • Precursors – Carbomyl phosphate & Aspartate • De novo synthesis - in the Liver cytosol • Energetic: Requires 2ATP ATP, PRPP Carbamoyl phosphate synthetase II (gln) Glutamine + 2ATP + CO2 phosphate - UDP UTP Glu + Pi + ADP CTP Aspartate Carbamoyl transcarbamoylase Glu + Pi + 2 ADP Gln + ATP Carbamoyl -aspartate Aspartate Orotate +2 ADP UTP +2 ATP Orotate phosphoribosyl transferase UMP +PRPP OMP Orotidylic acid decarboxylase Figure1 - Biosynthesis of the Pyrimidine nucleotides UTP & CTP Regulation of Pyrimidine Biosynthesis CPS-II Asp. Trans carbomylase OMP Decarboxylase CATABOLISM OF PYRIMIDINE NUCLEOTIDES • Pyrimidine degraded in the liver. • The end products - are nitrogenous bases Cytosine, Uracil and Thymine. • The bases are then degraded to amino acids, namely β-Alanine (from Cytosine & Uracil) & β-Amino isobutyrate (from Thymine). • These amino acids undergo transamination & other reactions to finally produce Acetyl coA, Succinate & CO2. • Clinical significance: β-Aminoisobutyrate is excreted in large quantity in Leukemia & when body subjected to X-ray irradiation. 2. Reye’s syndrome • Secondary Oratic aciduria • Defect in Ornithine transcarbamoylase (in urea cycle) Causes • Accumulation of Carbomyl phosphate • This is then diverted for the increased synthesis and excretion of oratic acid. Clinical significances of pyrimidine metabolism Oraticaciduria • Rare inherited disorders. • Enzyme deficiency- Orotate phosphoribosyl transferase and OMP decarboxylase. • Defect - in UMP formation & thus ↑ Oratate synthesis in blood & its excretion in the urine. • Causes - Growth retardation, severe anemia caused by hypo chromic erythrocytes and megaloblastic bone marrow. Leukopenia is also common. • Treatment – Diet rich in uridine & cytidine, which ↑UMP production via the action of nucleoside kinases. UMP then inhibits CPS-II, thus ↓ orotic acid production.