Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Circular dichroism wikipedia , lookup
Rosetta@home wikipedia , lookup
Structural alignment wikipedia , lookup
List of types of proteins wikipedia , lookup
Protein design wikipedia , lookup
P-type ATPase wikipedia , lookup
Protein folding wikipedia , lookup
Bimolecular fluorescence complementation wikipedia , lookup
Homology modeling wikipedia , lookup
Intrinsically disordered proteins wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Protein structure prediction wikipedia , lookup
Western blot wikipedia , lookup
Protein moonlighting wikipedia , lookup
Protein mass spectrometry wikipedia , lookup
Nuclear magnetic resonance spectroscopy of proteins wikipedia , lookup
Protein purification wikipedia , lookup
Trimeric autotransporter adhesin wikipedia , lookup
Predicting Function
(& location & post-tln modifications)
from Protein Sequences
June 15, 2015
Outline
Usefulness of protein domain analysis
Types of protein domain databases
Interpro scan of multiple domain DB
Using the SMART database
Predicting post-translational modifications
When annotation is NOT enough
You’ve got a list of genes, most of which have
been annotated with gene ontology and a
potential protein function
Why would you want to go on and look more
specifically at the protein domains?
Limitations of annotation
Even in a model organism with large amount of
resources, most genes are still annotated by
similarity
Often, the name given is based on the BEST match
to a particular domain or known protein
But…
Limitations of BLAST
Likelihood of finding a homolog to a sequence:
>80% bacteria
>70% yeast
~60% animal
Rest are truly novel sequences
~900/6500 proteins in yeast without a known
function
NAME: Similar to yeast protein YAL7400 not very
informative
Limitations of similarity
Proteins with more than one domain cause
problems.
Numerous matches to one domain can mask
matches to other domains.
Increased size of protein databases
Number related sequences rises and less related
sequence hits may be lost
Low-complexity regions can mask domain
matches
Proteins are modular
Individual domains can and often do fold
independently of other domains within the same
protein
Domains can function as an independent unit (or
truncation experiments would never work)
Thus identity of ALL protein domains within a
sequence can provide further clues about their
function
Proteins can have >1 domain
The name: protein kinase receptor UFO doesn’t
necessarily tell you that this protein also contains IgG and
fibronectin domains or that it has a transmembrane
domain
Domains are not always functional
If a critical residue is
missing in an active site, it’s
not likely to be functional
A similarity score won’t
pick that up
Multiple protein domain databases
Protein signature databases
Identify domains or classify proteins into families to
allow inference of function
Approaches include:
regular expressions and profiles
position-specific scoring matrix-based fingerprints
automated sequence clustering
Hidden Markov Models (HMMs)
PROSITE
Regular expression patterns describing functional
motifs
M-x-G-x(3)-[IV]2-x(2)-{FWY}
Enzyme catalytic
sites
Prosthetic group attachment sites
Ligand or metal binding sites
Either matches or not
Some families/domains defined by co-occurrence
Citrate synthase
G-[FYAV]-[GA]-H-x-[IV]-x(1,2)-[RKTQ]-x(2)-[DV]-[PS]-R
PRINTS
Similar to PROSITE patterns
Multiple-motif approach using either identity or
weight-matrix as basis
Groups of conserved motif provide diagnostic
protein family signatures
Can be created at super-family, family and sub-
family level
http://www.bioinf.manchester.ac.uk/dbbrowser/PRINTS/index.php
Profile-HMMs
Models generated from alignments of many homologues then
counting frequency of occurrence for each amino acid in each
column of the alignment (profile).
Profile-HMMs used to create probabilities of occurrence
against background evolutionary model that accounts for
possible substitutions.
Provides convenient and powerful way of identifying
homology between sequences.
Find domains in sequences that would never be found by
BLAST alone
HMM domain databases
Pfam
Classify novel sequences into protein domain profiles
Most comprehensive; >13,000 protein families (v26)
SMART
Signaling, extracellular and chromatin proteins
Identification of catalytic site conservation for enzymes
TIGRFAMs
Families of proteins from prokaryotes
PANTHER
Classification based on function using literature evidence
PFAM
>16,230 manually curated profiles
Can use the profile to search a genome for
matches
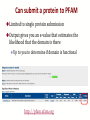
Can submit a protein to PFAM
Limited to single protein submission
Output gives you an e-value that estimates the
likelihood that the domain is there
Up to you to determine if domain is functional
http://pfam.xfam.org
Keyword search
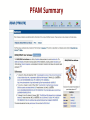
PFAM Summary
PFAM Domain Organization
PFAM Interactions
SMART database
SMART: Simple Modular Architecture Research Tool
Focus on signaling, extracellular and chromatin-
associated proteins
Curated models for >1200 domains
Use?
I have several kinase domains in my protein list and
want to know which ones are functional.
What other domains are found in signaling proteins?
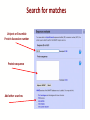
Search for matches
Uniprot or Ensemble
Protein Accession number
Protein sequence
Add other searches
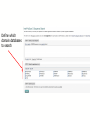
SMART Output
Mouse over for
information
Prediction of FUNCTIONAL catalytic activity
Can browse the domains
InterPro Scan
Combines search methods from several protein
databases
Uses tools provided by member databases
Uses threshold scores for profiles & motifs
Interpro convenient means of deriving a
consensus among signature methods
Define which
domain databases
to search
Example InterProScan search
Submitting an olfactory receptor gene (member
of the GPCR class of proteins) to InterPro
InterPro
family
2nd
InterPro
family
Submitting a different human GPCR protein to
Interpro
Same
InterPro
family
New
InterPro
family
InterProScan Families
InterProScan annotation
SMART & PFAM search
SMART DB results:
PFAM DB results:
Are 2 proteins homologs?
S. cerevisiae Ste3 is a GPCR pheromone receptor
Similarity to C. gatti protein:
25% identical, 45% similar, E-value 10-25
Very similar domain content and
arrangement
Advantage of InterProScan
Interpro integrates the different databases to
create a protein family signature.
Pfam/SMART/PANTHER/Gene3D & TIGR-FAM
will find domain families
PROSITE can find very specific signature patterns
PRINTS can distinguish related members of same
protein family
Cannot change the statistical cut-off for what is considered a
significant match
Function from sequence
Membrane bound or secreted?
GPI anchored?
Cellular localization?
Post-translational modification sites?
CBS prediction services
Protein sorting
SignalP, TargetP, others
Post-translational modification
Acetylation, phosphorylation, glycosylation
Immunological features
Epitopes, MHC allele binding, ect
Protein function & structure
Transmembrane domains, co-evolving positions
Transmembrane domain prediction
Phosphorylation prediction
O-glycosylation
EMBOSS
Open source software for molecular biology
Predict antigenic sites
Useful if want to design a peptide antibody
Look for specific motifs, even degenerate
Known phosphorylation motifs
Find motifs in multiple sequences with one
submission
Get stats on proteins/nucleic acid sequences
Sequence manipulation of all kinds
Today in lab
Tutorial on protein information sites
From a sublist generated using DAVID, generate a
list of protein IDs and obtain the sequences
Obtain protein accession numbers for the cluster
Submit to SMART database to
characterize/analyze the domains
Pick 2 proteins to do additional predictions