Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Extrachromosomal DNA wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Protein moonlighting wikipedia , lookup
Microevolution wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Metabolic network modelling wikipedia , lookup
Tay–Sachs disease wikipedia , lookup
Expanded genetic code wikipedia , lookup
Oncogenomics wikipedia , lookup
Genetic code wikipedia , lookup
Frameshift mutation wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Epigenetics of neurodegenerative diseases wikipedia , lookup
Medical genetics wikipedia , lookup
Neuronal ceroid lipofuscinosis wikipedia , lookup
Mitochondrial DNA wikipedia , lookup
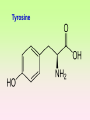
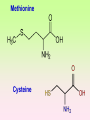
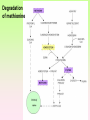
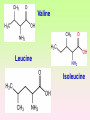
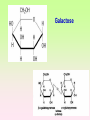
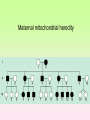
Enzymopathy – Inherited Metabolic Disorders RNDr. Hana Zoubková, PhD Energy -228 substrate Mutation in nuclear DNA Dysfunctional enzyme, protein product Multi-organ failure Mutation in mitochondrial DNA General characteristics • Inherited metabolic disorders caused by enzyme disorders • hereditary type recessive – Heterozygotes Aa with 50% residual activity of enzyme are clinically normal. • diffused versus macromolecular substrates – Macromolecular s. are only in the tissue, where the substrate is accumulated. • acumulation of substrate (1), deficit of product (2) of origin of toxic substances, (3) or combination General consequences 1. the accumulation of molecules proteins, enzyme, which is not eliminated and excluded in the right way 2. the absence of the protein molecules, enzyme or other substances in the organism 3. the formation of by-substances (proteins, enzymes, or other substances), which do not belong to the body, and cause acute or slow poisoning General characteristics • A single patient may have a loss of more than one enzyme. They share the same cofactor, a common activator, modifying or stabilizing protein; may be missing a whole group of enzymes or the entire organelle is abnormal. • phenotype homology Diseases are caused by other enzymes, but in the same area of metabolism. The diseases are different, but arise in the partial or complete defect of one enzyme. • It does not concern catalytic RNAs. General characteristics • It is currently recognized more than 850 IMD. Approximately 100 of them are curable or controllable by diet. • Low-protein, lactose free, gluten free diet Every year is born around 1,000 children with IMD in Czech republic Inherited metabolic disorders IMD aminoacids: Hyperphenylalaninemia, Tyrosinemia, Alkaptonuria, Homocystinuria, Cystinuria, Cystinosis Leucinosis - Organic aciduria (acidemia) IMD saccharides: Galactosemia, Galactokinase deficiency, Fruktose-intolerance, UDP-Galactose-4-epimerase deficiency, Fructose 1,6-Diphosphatase deficiency, glykogenosis IMD purin/pyrmidin - Porfyria, disorders of urea cycle IMD lysosoms - Tay-Sachs disease peroxisoms, mitochondria Tyrosinemia, new type acid oxidase Fumarylacetoacetate hydrolase block Tyrosinemia, type I Hyperphenylalaninemia Is characterized by mildly or strongly elevated levels of the aromatic amino acid phenylalanine in the blood. Are caused by lossing mutations in gene for enzyme phenylalanine hydroxylase (PAH) or in gene for cofactor. Enzyme is necessary for metabolism of amino acid phenylalanine (Phe) to the amino acid tyrosine. Phenylketonuria - PKU Variant PKU Non-PKU Phenylketonuria PKU AA phenylalanine accumulates and is converted into phenylpyruvate (phenylketone), which is detected in the urine. It is toxic for the body and causes mental retardation, light pigmentation. Main treatment for classic PKU patients is a strict PHE-restricted diet. chromosome 12q24.1, incidence 1:5 000 a 1:15 000 1:6 000 in Cz Alelic heterogenity - 400 alleles, 6 mutations in each of the 13 exons Newborn screening od PKU is included in the panel of most countries. Treatment is the diet – it is effective, if it is launched at birth. Tyrosine Tyrosinemia, new type acid oxidase Fumarylacetoacetate hydrolase block Tyrosinemia, type I Albinism classical, type IA. • lack of product, pigment of skin and hair, achromatosis • lack or non-functional tyrosinase Tyrozinase negative • Typ IB – tyrozinse is positive, non- active • Type II – frequent, tyrozinase positive but with gene mutation Alkaptonuria - AKU the failure of metabolism of homogentisic acid, Caused by lack of enzyme homogentisate 1,2dioxygenase, homogentisic acid oxidase (HGD), Accumulation of substrate Homogentisic acid is changed to brown-black pigment – alkapton - excretion to the urine, accumulation in tissues ochronosis incidence 1 : 250 000 Tyrosinemia type I is caused by the failure of tyrosine metabolism, of enzyme fumarylacetoacetate hydrolase (FAH). Accumulation of tyrosine (substrate) and toxic secondary metabolites, mainly for the liver and kidneys Incidence 1: 100 000-120 000 Tyrosinemia type II and III are rare and the level of toxic metabolite sukcinylaceton is not so high. Treatment of alkaptonuria and tyrosinemia by diet and nitison. Methionine Cysteine Degradation of methionine Homocystinuria Methionine is metabolized to cysteine via homocysteine. Is caused by the lack of cystathionine β-synthase (CBS), which causes the increased value of homocysteine and methionine in the urine. Accumulation of substrate is harmful for four organ systems: the eye, skeleton, vascular endothelium and central nervous system Ectopia lensis, excessive height, length of limbs, vascular abnormalities Valine Leucine Isoleucine Organic aciduria (acidemia) The group of specific enzymes disorders in the catabolism of branched amino acids leucin, valin and isoleucin. Accumulation of toxic organic acids (by-products) causes medium acidification of organism, especially disability of brain. Every year is born 10 patients with one of the organic aciduria in Czech Republic „Maple syrup urine disease - MSUD“ Or Leucinosis Is the failure of metabolism of branched-chain α-keto acids. The failure of certain dehydrogenase in multienzyme complex Accumulation of keto-acids in the body, accumulation of substrate causes food intolerance, failure to thrive, vomiting, lethargy, and the smell of urine and earwax after maple syrup. 1: 20 000-50 000, Mennonites – 1: 1000 Galactose Galactosemia, classical Is the failure of metabolism of monosaccharide galactose (component of lactose). Toxic by-products of metabolism are toxic to liver, brain, kidneys and eye lens; cause malnutrition, cirrhosis and mental retardation. Galactose-1-phosphate uridyltransferase – GALT (9.chromosome, 160 mutations) 1:18 000 to 1:70 000 Tay-Sachs disease GM2–gangliosidosis Is the failure of degradation of sfingolipid GM2 – gangliosid. It is the hexosaminidase A (HEXA) deficiency, which causes accumulation of substrate. Substrate affects mainly the brain and causes degeneration of nerve system. Syndrome - cherry-red spot on retina increased incidence (1:3600) for ashkenazi Jewish population (1:360 000, chromosome 15) Porfyria Caused by enzymes deficiencies operating in the synthesis of heme. Defects of enzyme in the early stage cause accumulation of substrate: acid 5aminolevulinic acid and porfobilinogen Defects in the later stages lead to the accumulation of porfyrinogens - accumulation of substrate and byproducts Skin, liver damage congenital erytropoetic porfyria Nuclear and mitochondrial heredity Mutated gene in nucleus Mutated gene v mitochondria Combination of maternal and Genetic maternal information paternal genetic information of prokaryotic type - chromosomes Mendel‘s law Maternal line Disability throughout the body Variable expression Maternal mitochondrial heredity Mitochondrial diseases are a heterogeneous group with dysfunctions in respiratory chain. They are metabolic disorders and neurodegenerative or muscle diseases. Are caused by the lack of product. Affection is multi-systematic, with exception of LHON Tissue with a high level of requirement of ATP (the brain, muscle, liver, heart, kidney . . .) are affected. Mutations in mtDNA Variable expresivity typical for mitochondrial disease - distribution to daughter cells is random, distribution of mutated and normal mtDNA is variable Homoplasmy = only mutated or only normal mtDNA in the cell the mixture of normal and mutant mtDNA = Heteroplasmy Defect in mitochondrial respiratory chain should be considered for patients, which have any unexplainable combination of neuromuscular and/or different symptoms, with proceeding course and affecting seemingly unrelated organs. Munnich et al, OMMBD, chapter 99 Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) - brain and nervous system (encephalo-) and muscles (myopathy), lactic acid, muscle spasms, impaired muscle coordination (ataxia) Leber hereditary optic neuropathy – LHON degeneration of retinal ganglion cells, acute or subacute loss of central vision MELAS, Association with mutaion (A3243G) mtDNA in gene for tRNA Leu (UUR). Mutations in mtDNA Mutations cca 10x more often then in nuclear DNA, because histons and some reparation mechanisms are absent. Approximately 10% of all mitochondrial proteins are encoded by nucleus. In case of mutations in these genes - AR heredity Some mitochondrial proteins are aggregates with origin in nucleus and in mitochondria. Thanks for attention Literature Thompson and Thompson: Clinical Genetics, 6. edition, 2004 Adkinson J R, Brown M.D.: Elsevier‘s Integrated Genetics, 2007