Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Paracrine signalling wikipedia , lookup
Silencer (genetics) wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Genetic code wikipedia , lookup
Gene expression wikipedia , lookup
Expression vector wikipedia , lookup
Magnesium transporter wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Point mutation wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
Bimolecular fluorescence complementation wikipedia , lookup
Biochemistry wikipedia , lookup
Metalloprotein wikipedia , lookup
Interactome wikipedia , lookup
Protein purification wikipedia , lookup
Western blot wikipedia , lookup
Structural alignment wikipedia , lookup
Two-hybrid screening wikipedia , lookup
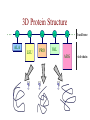
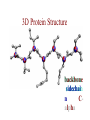
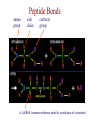
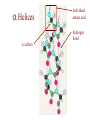
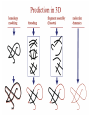
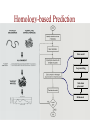
Protein Modeling Protein Structure Prediction 3D Protein Structure … Cα Cα Cα Cα LEU PRO VAL ? ? ALA Cα … ARG ? backbone sidechain The Protein Folding Problem • we know that the function of a protein is determined in large part by its 3D shape (fold, conformation) • can we predict the 3D shape of a protein given only its amino-acid sequence? Motivation • Want to identify the function of genes we find, and what different mutations/alleles do • One gene = one protein (sort of) – Function of protein = function of gene • Function can be determined in many ways – Gene expression, knockouts, etc – But these take time, and are prone to mistakes • Goal: If we can structure every protein, learning their functions isn’t too far away Thornton et al 2000 (Nature) Protein Architecture • proteins are polymers consisting of amino acids linked by peptide bonds • each amino acid consists of – a central carbon atom – an amino group NH 2 – a carboxyl group COOH – a side chain • differences in side chains distinguish different amino acids 3D Protein Structure baacckkbboonnee sidechhaaiin n Calpha Peptide Bonds amino group side chain carboxyl group carbon (common reference point for coordinates of a structure) Amino Acid Side Chains • side chains vary in: shape, size, charge, polarity Levels of Description • protein structure is often described at four different scales – primary structure – secondary structure – tertiary structure – quaternary structure Levels of Description Secondary Structure • secondary structure refers to certain common repeating structures • it is a “local” description of structure • two common secondary structures helices strands/sheets • a third category, called coil or loop, refers to everything else Helices carbon individual amino acid hydrogen bond Sheets Ribbon Diagram Showing Secondary Structures Levels of Description What Determines Conformation? • in general, the amino-acid sequence of a protein determines the 3D shape of a protein [Anfinsen et al., 1950s] • but some exceptions – all proteins can be denatured – some proteins are inherently disordered (i.e. lack a regular structure) – some proteins get folding help from chaperones – there are various mechanisms through which the conformation of a protein can be changed in vivo – post-translational modifications such as phosphorylation – prions – etc. What Determines Conformation? • what physical properties of the protein determine its fold? – rigidity of the protein backbone – interactions among amino acids, including • electrostatic interactions • van der Waals forces • volume constraints • hydrogen, disulfide bonds – interactions of amino acids with water Determining Protein Structures • protein structures can be determined experimentally (in many cases) by – x-ray crystallography – nuclear magnetic resonance (NMR) Myoglobin From www.inst.bnl.gov/GasDetectorLab/x-rays/SRI94.htm Myoglobin S.E.V. Phillips. "Structure and refinement of oxymyoglobin at 1.6 Å resolution.", J. Mol. Biol. 1980, 142, 531. X-ray Crystallography x-ray beam protein crystal collection plate diffraction pattern electron density map (“3D picture”) Electron Density Map Interpretation … … GIVEN: 3D Electron Density Map Electron Density Map Interpretation … … FIND: All-atom Protein Model NMR • Nuclear Magnetic Resonance Spectroscopy • Cannot handle large proteins like X-ray • Exploits the chemical environment to return distances between atoms – Can use knowledge of restraints to identify positions of atoms that produce peaks Protein structure determination in solution by NMR spectroscopy Wuthrich K. J Biol Chem. 1990 December 25;265(36):22059-62 Experimental Methods • Very expensive and time-consuming – Computational methods can help with time • Many proteins still cannot be done in this manner More motivation • there is a large sequence-structure gap ≈300K protein sequences in SwissProt database ≈50K protein structures in PDB database • key question: can we predict structures by computational means instead? Approaches to Protein Structure Prediction • prediction in 1D – secondary structure – solvent accessibility (which residues are exposed to water, which are buried) – transmembrane helices (which residues span membranes) • prediction in 2D – inter-residue/strand contacts • prediction in 3D – homology modeling – fold recognition (e.g. via threading) – ab initio prediction (e.g. via molecular dynamics) Prediction in 1D, 2D and 3D predicted secondary structure and solvent accessibility known secondary structure (E = beta strand) and solvent accessibility Figure from B. Rost, “Protein Structure in 1D, 2D, and 3D”, The Encyclopaedia of Computational Chemistry, 1998 2D Prediction Approaches • use secondary structure predictions to predict short-range contacts (e.g. hydrogen bonds in α helices) • use secondary structure predictions to predict β strand alignments • use correlated mutations to predict contacts Prediction in 3D • homology modeling given: a query sequence Q, a database of protein structures do: • find protein P has high sequence similarity to Q • return P’s structure as an approximation to Q’s structure • fold recognition (threading) given: a query sequence Q, a database of known folds do: • find fold F such that Q can be aligned with F in a highly compatible manner • return F as an approximation to Q’s structure Prediction in 3D • fragment assembly(Rosetta) given: a query sequence Q, a database of structure fragments do: • find a set of fragments that Q can be aligned with in a highly compatible manner • return the combined fragments as an approximation • molecular dynamics given: a query sequence Q do: • use laws of physics to to simulate folding of Q Homology Modeling • most pairs of proteins with similar structure are remote homologs (< 25% sequence identity) • homology modeling usually doesn’t work for remote homologs ; most pairs of proteins with < 25% sequence identity are unrelated probably unrelated 0% remote homologs homologs 20% 30% pairwise sequence identity 100% Homology-based Prediction Raw model Loop modeling Side chain placement Refinement The SCOP Database Structural Classification Of Proteins FAMILY: proteins that are >30% similar, or >15% similar and have similar known structure/function SUPERFAMILY: proteins whose families have some sequence and function/structure similarity suggesting a common evolutionary origin COMMON FOLD: superfamilies that have same secondary structures in same arrangement, probably resulting by physics and chemistry Examples of Fold Classes Threading Ab initio Prediction – ROSETTA 1. PSI-BLAST – homology search Discard sequences with >25% homology 2. PHD For each 3-long and each 9-long sequence fragment, get 25 structure fragments that match “well” ? ? ? ai.stanford.edu/~serafim/CS262_2006/Slides Ab initio Prediction – CASP results Summary of current state of the art Open Ended • • • • • • Ab Initio is the goal, far from it Sidechain prediction Contact Map prediction Search space reduction Parallelization (GPUs) Surface Accessibility Other areas • • • • • Protein-Protein Interaction Drug Design Protein Engineering Ligand Docking/Inhibition Function Prediction