Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Epigenetics in stem-cell differentiation wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
DNA supercoil wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
DNA damage theory of aging wikipedia , lookup
Molecular cloning wikipedia , lookup
Primary transcript wikipedia , lookup
DNA vaccination wikipedia , lookup
Deoxyribozyme wikipedia , lookup
Cancer epigenetics wikipedia , lookup
History of genetic engineering wikipedia , lookup
Point mutation wikipedia , lookup
Cre-Lox recombination wikipedia , lookup
Extrachromosomal DNA wikipedia , lookup
Oncogenomics wikipedia , lookup
Mir-92 microRNA precursor family wikipedia , lookup
Polycomb Group Proteins and Cancer wikipedia , lookup
Cancer Research UK Scientific Yearbook 2002-03
Studying DNA replication to find
smarter cancer drugs
Basic research into DNA replication, a
central part of the cell division cycle, has
Underreplication:
revealed new drug targets with potential
for selective killing of cancer cells.
Before a cell can successfully divide
to produce two daughter cells, it
must precisely duplicate its
chromosomal DNA. This process,
DNA replication, is targeted by several
‘antimetabolite’ cancer drugs such as
methotrexate and fluorouracil that
reduce the supply of deoxynucleotide
precursors. But normal cells as well as
cancer cells have to replicate their
DNA, so inhibitors of DNA
replication should indiscriminately kill
all dividing cells and have the
anticancer specificity of a hand
grenade. How come they work?
Years ago, when I was a wayward
medical student, the specificity of the
antimetabolites was explained in terms
of cancer cells dividing more
frequently than normal cells. From a
modern perspective, the real answer
seems likely to be much more
complex. We now know that cells
possess intricate feedback networks,
termed ‘checkpoints’. These monitor
the success of different cell cycle tasks
and provide remedial action, or block
further cell cycle progress should
problems be detected. Most cancer
cells show defects in one or more
checkpoint pathways, and it seems
likely that this accounts for their
decreased tolerance to a disruption of
their normal supply of
deoxynucleotides by antimetabolites.
My lab is funded by Cancer Research
UK to study the way that
chromosomal DNA replication is
controlled. Through this research we
are beginning to identify new potential
drug targets whose inhibition promises
40
Nomal
replication:
Overreplication:
G1
S
G2
M
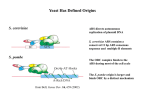
Figure 1: A small segment of chromosomal DNA, replicated from three origins is shown during the cell cycle.
Middle panel: successful duplication. Top panel: under replication due to the failure of one of the origins to fire.
As sister chromatids are separated during anaphase, the chromosome is likely to break near the unreplicated
section. Bottom panel: over-replication, due to one of the origins firing a second time in S phase. The local
duplication of DNA in the vicinity of the over-firing origin is likely to represent an irreversible genetic change, and
might be resolved to form a tandem duplication.
greater specificity for cancer cell killing
than is achieved by the old
antimetabolites.
result will be the localised rereplication of DNA in the vicinity of
the origin (lower lane of Figure 1).
The replication licensing system
During S phase of the cell division
cycle, pairs of replication forks are
initiated ('fired') at thousands of
replication origins scattered
throughout the genome. The
replication forks are protein machines
that move along double-stranded
DNA replicating both strands as they
go. The two forks initiated at each
origin move away from each other
along the DNA, creating a bubble of
replicated DNA (coloured blue in
Figure 1). They terminate when they
encounter a replication fork coming
from the opposite direction, which
happens when all the DNA between
adjacent origins has been replicated
(middle lane of Figure 1).
As a Cancer Research Campaignfunded graduate student with Professor
Ron Laskey, I obtained results
suggesting that cells minimise these
dangers by separating the process of
DNA replication into two distinct
phases. In the first phase, proteins are
loaded onto replication origins to
‘license’ them for a single initiation
event. The licence is essential for
initiation to occur, but is displaced from
origins as the DNA replicates. By
ensuring that the licensing system is
shut down before S phase starts, origins
will fire just once in each cell cycle.
The outcome should be the precise
duplication of the genomic DNA. But
to achieve this, the cell has to regulate
its replication origins very strictly. If too
few origins are used, then some of the
intervening DNA may be left
unreplicated (upper lane of Figure 1).
In contrast, should an origin fire more
than once in a single cell cycle, the
Several years ago, my lab (this time
funded by Imperial Cancer Research
Fund) developed one of the first
assays for components of the
replication licensing system. We have
used this assay to undertake a
systematic purification of all the
activities required to assemble licensed
replication origins on a simple
chromatin template. Along with the
labs of Laskey and Dr Haruhiko
Takisawa, we showed that the
replication licence itself was provided
Cancer Research UK Scientific Yearbook 2002-03
Licensing is strictly regulated
The licensing system becomes active
during late mitosis, just as a new cell is
born (Figure 2). In order to prevent
over-replication of DNA, the ability to
license new origins must be shut down
before replication starts – there is no
late licensing in the cell cycle bar! It is
known from work first carried out by
Sir Paul Nurse and colleagues, that
cyclin-dependent kinases (CDKs) play
an important role in suppressing relicensing of replicated DNA. CDKs are
activated in late G1 and drive the cell
through S phase and mitosis. They are
inactive in late mitosis and early G1,
thus providing a time window in the
cell cycle for the licensing system to be
active. A variety of results suggested,
however, that there must be other
activities that inhibit the licensing system.
The existence of redundant controls
(belt and braces) is perhaps not
surprising, given the dire consequences
that re-replication would have to the
cell. We have recently shown that a
small protein called Geminin binds and
inhibits Cdt1/RLF-B during S phase, G2
and mitosis, and plays a major part in
Licensing
M
M
ORC
M
M
M
Cdt1
Cdc6
ORC
M
M
G2
G1
M
RLS
by a complex of the six
minichromosome maintenance
proteins, Mcm2-7 (Figure 2). Further
work identified several other proteins
that are required to load Mcm2-7
onto DNA. Together these proteins
form the ‘pre-replicative complex’
(pre-RC) identified by Dr John Diffley,
another Cancer Research UK-funded
scientist and close colleague. First, the
origin recognition complex (ORC)
binds DNA to define where the
replication origin will be positioned.
ORC then recruits two further
proteins, Cdc6 and Cdt1/RLF-B. Acting
in concert, these three proteins then
recruit multiple copies of the Mcm2-7
hexamer to the origin DNA, thereby
licensing it. Once this licensing reaction
is complete, neither ORC, Cdc6 nor
Cdt1/RLF-B are required to maintain
Mcm2-7 on the DNA. The licensing
system can be shut down at the end
of G1 by inactivation of ORC, Cdc6
or Cdt1/RLF-B without affecting the
Mcm2-7 already bound to the origin.
Cdt1
Cdc6
ORC
M
S
M
M
M
M
M
M
Cdt1
Cdc6
ORC
M
M
M
RLS
M
M
Free Mcm(2-7) Replication origin
Replication licensing
proteins
licensed by Mcm(2-7) system active
Figure 2: A small segment of DNA containing three replication origins is shown during the cell division cycle. As a new cell is born during late
mitosis (M phase), the replication licensing system is activated and origins become licensed by loading Mcm2-7 to form a pre-replicative
complex. During G1 the cell awaits signals that it is appropriate to undergo a further round of cell division. It then enters S phase, when the
DNA is replicated. As replication forks are initiated at licensed origins, Mcm2-7 is displaced from the origins. The licensing system is inactive
and Mcm2-7 cannot be re-loaded onto replicated origins. By G2 all the DNA is replicated. In mitosis (M phase) the DNA condenses into
chromosomes, which are divided and segregated to the two new daughter cells.
inactivating the licensing system at
these stages. Geminin activity is
downregulated in late mitosis and G1
by at least two mechanisms. Protein
levels are kept low by cell cycle
regulated proteolysis, while the
remaining protein is inactivated to
prevent it interacting with Cdt1/RLF-B.
Understanding how Geminin activity is
regulated is a major research objective
over the next few years.
unlicensed, the unbound Mcm2-7
being degraded (Figure 3). Cells that
have only temporarily withdrawn from
the cell cycle into the ‘G0’ state must
then re-license their origins if they are
stimulated to divide again. The
unlicensed state of quiescent cells may
ensure that they don’t re-enter the cell
cycle inappropriately. Cancer cells
usually maintain a high proliferation
capacity, and so maintain high Mcm2-7
levels. Laskey and Professor Gareth
Williams (a Cancer Research UK
Senior Clinical Research Fellow) are
exploiting this feature to develop new
diagnostic tests for cancer.
In a further twist to the licensing story,
licensed G1 cells that withdraw from
cell proliferation subsequently lose their
origin-bound Mcm2-7. They become
Late
mitosis
M
M
M
M
M
M
M
M
M
S
phase
G1
M
M
M
M
M
De-licensing
Permanent withdrawl
from proliferation
M
Re-licensing
G0
Figure 3: G1 cells can stop proliferating and withdraw into the G0 state. As they do this, Mcm2-7 is displaced
from the origins and degraded (‘de-licensing’). When G0 cells are stimulated to start proliferating again, they must
re-synthesize Mcm2-7 and re-load them onto replication origins (‘re-licensing’).
41
Cancer Research UK Scientific Yearbook 2002-03
Cell cycle
phase
Late G1
S phase
Trigger
DNA damage
sufficient licensing ?
DNA damage
stalled replication forks
Effect
Prevent entry
into S phase
Prevent further
origin firing
Figure 4: Checkpoints affecting S phase
A
B
Figure 5A: Normal cells overexpressing Geminin.
Figure 5B: Cancer cells overexpressing Geminin. The DNA in the nucleus is labelled
blue, and an irregular shape indicates cell death. The cells are stained with DAPI
The consequence of licensing
inhibition in normal and cancer cells
Checkpoints ensure that the cell
embarks on cell cycle processes only
when conditions are appropriate. For
example, a G1 checkpoint blocks entry
into S phase if DNA is damaged, while
an intra-S checkpoint blocks the
initiation of new replication forks if
existing forks have stalled (Figure 4).
The combined activity of CDKs and
Geminin ensure that once the cell has
entered S phase, further origin
licensing is prohibited. At the start of S
phase, the cell must therefore have a
sufficient number of licensed origins to
complete the job.
Before you set out on a journey to
cross uninhabited moors, you check
that you have enough petrol to get
across. Does the cell do the same? Is
there a ‘licensing checkpoint’ that
delays entry into S phase until enough
origins are licensed? To address this
question, I teamed up with Sir David
Lane at the nearby Medical School in
Dundee to exploit his knowledge of
cell cycle checkpoints. Together we set
a graduate student, S. Shreeram, onto
the problem. Since Geminin is
42
currently the only known specific
inhibitor of licensing, we decided to
force cells to overexpress a
constitutively active form of Geminin
using an adenoviral delivery system.
Shreeram first showed that this virallydelivered Geminin severely reduced
the ability of a variety of human cell
lines to load Mcm2-7 onto DNA. Not
surprisingly, this caused them all to
stop proliferating. But on closer
inspection, different cell lines showed
different responses to this inhibition of
licensing activity. Normal (primary)
human cells over-expressing Geminin
showed all the features of cells
arrested in G1, patiently waiting
without attempting to start DNA
replication. There were no signs of
DNA replication having started, and
checkpoint signals indicative of
replication problems were not present.
CDK activity was typical of cells in late
G1. These features suggest that the
cells possess a ‘licensing checkpoint’
preventing them from entering an S
phase that they have too few licensed
origins to complete (Figure 5A). It
would be reasonable to expect that,
were the virally delivered Geminin
withdrawn, the cells would be in a cell
cycle state where they could complete
origin licensing before embarking on S
phase itself. More sophisticated
experiments are required to
determine this.
None of the cancer cell lines
Shreeram looked at behaved in this
orderly fashion. All of them showed
clear signs of being stuck in an S phase
that they could not complete, and
checkpoint signals indicative of
replication problems were strongly
induced. Most dramatically, all the
cancer cells ultimately underwent
programmed cell death (apoptosis)
as a consequence of Geminin
overexpression (Figure 5B). Even if
the virally delivered Geminin were
withdrawn from these cells when
they were still alive, they would never
be able to recover because of the
normal control systems that prevent
re-licensing of origins once S phase
has started. These cancer cells
therefore appear to have lost the
‘licensing checkpoint’, making them
uniquely sensitive to disruptions of
the licensing system.
Could this effect be the basis of a new
type of anticancer drug, showing a
much higher degree of cancer cell
selectivity than the old antimetabolites?
We certainly hope so. A 30 kDa
protein such as Geminin is, however,
unsuitable as a drug. We would need
a much smaller molecule that
interferes with replication licensing in a
similar way. We are currently
performing structure-function studies
of Geminin and Cdt1/RLF-B with the
ultimate aim of getting a threedimensional structure of their
interaction. However, the best way of
finding inhibitors still seems to be trial
and error – screening vast ‘libraries’ of
exotic chemicals to find one that has
the precise properties required.
Cancer Research UK’s New Targets
Committee has awarded us a oneyear grant to develop an assay suitable
for high-throughput screening for
Geminin-mimics. This ‘translational’
research is taking me into some
unfamiliar territory, but the new
challenges are stimulating. I hope this
will be the first of many new potential
anticancer targets that will arise from
studies on DNA replication.
Basic, translational and clinical
researchers must work together if we
are to produce new treatments that
kill cancer with the effectiveness we
require. Achieving this synergy is not
easy, and the creation of Cancer
Research UK can only help. Here in
Dundee, all Cancer Research UKfunded scientists have recently come
together to form a ‘co-operative
centre’ with the aim of stimulating
such interdisciplinary collaborations
and maximising potential. If we can
smooth the path from basic research
to anticancer drug creation, the future
looks bright.
J Julian Blow
Cancer Research UK Chromosome
Replication Research Group
University of Dundee