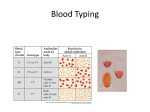
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Point mutation wikipedia , lookup
Human genome wikipedia , lookup
Gene expression programming wikipedia , lookup
Population genetics wikipedia , lookup
History of genetic engineering wikipedia , lookup
Neocentromere wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Y chromosome wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Heritability of IQ wikipedia , lookup
Ridge (biology) wikipedia , lookup
Public health genomics wikipedia , lookup
Minimal genome wikipedia , lookup
Biology and consumer behaviour wikipedia , lookup
Genomic imprinting wikipedia , lookup
Human genetic variation wikipedia , lookup
Gene expression profiling wikipedia , lookup
X-inactivation wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Genome evolution wikipedia , lookup
Designer baby wikipedia , lookup
Microevolution wikipedia , lookup
Brief Rapid Communication Genome-Wide Linkage Analyses of Systolic Blood Pressure Using Highly Discordant Siblings Julia Krushkal, PhD; Robert Ferrell, PhD; Stephen C. Mockrin, PhD; Stephen T. Turner, MD; Charles F. Sing, PhD; Eric Boerwinkle, PhD Downloaded from http://circ.ahajournals.org/ by guest on August 11, 2017 Background—Elevated blood pressure is a risk factor for cardiovascular, cerebrovascular, and renal diseases. Complex mechanisms of blood pressure regulation pose a challenge to identifying genetic factors that influence interindividual blood pressure variation in the population at large. Methods and Results—We performed a genome-wide linkage analysis of systolic blood pressure in humans using an efficient, highly discordant, full-sibling design. We identified 4 regions of the human genome that show statistical significant linkage to genes that influence interindividual systolic blood pressure variation (2p22.1 to 2p21, 5q33.3 to 5q34, 6q23.1 to 6q24.1, and 15q25.1 to 15q26.1). These regions contain a number of candidate genes that are involved in physiological mechanisms of blood pressure regulation. Conclusions—These results provide both novel information about genome regions in humans that influence interindividual blood pressure variation and a basis for identifying the contributing genes. Identification of the functional mutations in these genes may uncover novel mechanisms for blood pressure regulation and suggest new therapies and prevention strategies. (Circulation. 1999;99:1407-1410.) Key Words: hypertension n blood pressure n genetics n genes E levated blood pressure of unknown origin is a common risk factor for cardiovascular disease, stroke, end-stage renal disease, and peripheral vascular disease.1,2 The contributors to elevated blood pressure are poorly understood, but multiple lines of evidence have implicated both genetic and environmental factors.3 Studies aimed at identifying the genes that contribute to interindividual blood pressure variation have been limited to candidate genes; the results have proven informative but not always consistent.4 Investigations of several rare mendelian forms of high blood pressure, such as Liddle syndrome,5 have revealed causative mutations, but their very low frequency diminishes their public health impact. An alternative approach is to use genetic linkage analyses to systematically evaluate the human genome for segments that contain genes that influence blood pressure levels. Genome-wide genetic linkage studies for complex traits pose several challenges that require careful study design and analysis. Although these traits aggregate in families, they do not segregate in any clear mendelian fashion as a result of there being multiple contributing genes, each with polymorphic allele frequencies and small to moderate effects. In this report, we describe the results of using modern genome-wide linkage analysis methods and an efficient discordant sib pair design6,7 to localize genes that affect interindividual systolic blood pressure (SBP) variation. Methods These analyses were performed as part of the GENOA Network of the Family Blood Pressure Program and were based on data collected on 3974 members of 583 multigeneration pedigrees from Rochester, Minn, ascertained without regard to health status.8 SBP levels were measured for each individual 3 times $2 minutes apart with a random-zero sphygmomanometer, and the average of these 3 readings was used for the linkage analyses reported herein. Fifty-five pedigrees with 1 or more full siblings above the sex- and age-specific 80th percentile and 1 or more full siblings below the sex- and age-specific 20th percentile of the SBP distribution were identified from the sample of 583 pedigrees. These pedigrees contained 69 discordant full-sibling pairs. The two discordant groups of siblings were not significantly different (P.0.05) for average age (mean, 16 years), weight (mean, 57 kg), height (mean, 162 cm), and body mass index (mean, 21 kg/m2) or for gender prevalence (51% male). All 427 individuals in the 55 pedigrees were genotyped for 359 highly polymorphic marker loci located on the 22 human autosomes Received October 19, 1998; revision received December 22, 1998; accepted January 25, 1999. From the Institute of Molecular Medicine (J.K., E.B.) and the Human Genetics Center (E.B.), University of Texas–Houston Health Science Center, Houston, Tex; Department of Human Genetics (R.F.), University of Pittsburgh, Pittsburgh, Pa; National Heart, Lung, and Blood Institute (S.C.M.), Bethesda, Md; Division of Hypertension (S.T.T.), Department of Internal Medicine, Mayo Clinic, Rochester, Minn; and Department of Human Genetics (C.F.S.), University of Michigan, Ann Arbor, Mich. Guest Editor for this article was Joseph Loscalzo, MD, Boston University Medical Center, Boston, Mass. Detailed results of the genome-wide linkage analyses discussed in this article can be found on the World Wide Web at http://www.circulationaha.org Correspondence to Eric Boerwinkle, PhD, Human Genetics Center, University of Texas–Houston Health Science Center, PO Box 20334, Houston, TX 77225. E-mail [email protected] © 1999 American Heart Association, Inc. Circulation is available at http://www.circulationaha.org 1407 1408 Genome-Wide Analyses of Systolic Blood Pressure Significant and Suggestive Genomic Regions Containing Genes Affecting SBP Variation Lowest P Value of the Peak Nearby Markers t Chromosome ,0.0001 23.96 6 4 cM distal to D6S1009 0.0033 22.80 15 3 cM distal to D15S652 0.0076 22.49 5 At D5S1471 0.0089 22.43 2 At D2S1788 0.0160 22.19 18 At D18S843 0.0171 22.16 9 At D9S301 0.0193 22.11 2 2 cM distal to D2S441 0.0234 22.02 21 At D21S1446 0.0240 22.01 20 3 cM proximal to D20S478 0.0309 21.90 5 1 cM proximal to D5S817 0.0314 21.89 16 5 cM distal to D16S402 0.0353 21.84 2 At D2S1334 Significant P,0.01 Suggestive 0.01,P,0.05 Downloaded from http://circ.ahajournals.org/ by guest on August 11, 2017 (Research Genetics). Genotyping was performed by standard methods with an Applied Biosystems/Perkin Elmer 377 automatic DNA sequencer. Genotypes from all 427 individuals were used to calculate the multipoint identity by descent probabilities every 1 centimorgan (cM) by use of the hidden Markov model method.9 Marker order and genetic distances were provided by the CHLC/Weber screening set versions 8.0 and 6.0 (Research Genetics) and the Marshfield Medical Research Foundation (Marshfield, Wis). Genetic linkage between the marker and trait loci was assessed with the t statistic of Risch and Zhang.7 We compared identity by descent sharing among the 69 discordant sibling pairs with that expected under the null hypothesis of no linkage. Significance levels (probability values) were determined from a 1-sided Student’s t test with 68 degrees of freedom. Probability values ,0.01 were considered significant, and probability values between 0.01 and 0.05 were considered suggestive. The reader should be cautioned, however, that all results, significant or not, should be confirmed in a similar population before definite conclusions are drawn. Detailed results for each chromosome are not provided in this brief communication. Rather, they may be obtained on the World Wide Web (http://www.circulationaha.org) or by writing the communicating author (E.B.) Results Detailed results of the genome-wide linkage analyses are provided at http://www.circulationaha.org. Multipoint linkage analyses of the 22 autosomes identified 4 regions that showed significant linkage to genes that influence SBP variation (P,0.01) and 8 regions that showed suggestive linkage (0.01,P,0.05). The genomic location of these regions is described in the Table. The 12 regions are located on 9 chromosomes (chromosomes 2, 5, 6, 9, 15, 16, 18, 20, and 21). The 4 regions of particular interest by virtue of having probability values ,0.01 are located on chromosomes 2, 5, 6, and 15. The region with the greatest significance level rejecting the null hypothesis of no linkage between a marker locus and a gene influencing SBP variation was located on chromosome 6 (P50.00009; t523.96) between markers D6S1009 and D6S1003 (Figure, panel A). An interval 4 cM in length between positions 143 and 146 cM on chromosome 6 had a P value of #0.0001. A 12-cM interval on chromosome 6 between map positions 134 and 155 cM had P values ,0.01. On chromosome 15 at map position 97 cM from the tip of the short arm, the significance level was 0.0033 (t522.80), and the region between map positions 84 and 101 cM had a level of significance ,0.01 (panel B). At the distal end of the long arm of chromosome 5, the position of marker D5S1471 had a significance level of 0.0076 (t522.49) (panel C). Each position between 188 and 192 cM from the beginning of the chromosome 5 linkage map had a probability value ,0.01. On chromosome 2, the position of marker D2S1788 had a P value of 0.0089 (t522.43), and the map positions between 57 and 59 cM from the beginning of the chromosome 2 linkage map had a probability value ,0.01 (panel D). Discussion We report herein the results of the first genome-wide linkage analysis for genes that influence interindividual blood pressure variation in humans. This analysis identified several regions that are likely to contribute to genetic differences in SBP levels. Regions 2p22.1 to 2p21, 5q33.3 to 5q34, 6q23.1 to 6q24.1, and 15q25.1 to 15q26.1 on chromosomes 2, 5, 6, and 15, respectively, are significantly linked to genes that influence interindividual SBP differences. These regions contain a number of known genes, shown in the Figure, the products of which have previously been suggested to affect blood pressure regulation at the physiological level. These genes code for proteins involved in a variety of blood pressure– controlling systems, vascular smooth muscle tone, natriuresis, cellular calcium homeostasis, and hormonal and neural regulation of the renin-angiotensin system. For example, the product of the aminopeptidase N gene on chromosome 15 regulates vasopressin release via the mechanism of the N-terminal cleavage of angiotensin III.10 DNA sequence variants in these known genes and in nearby anonymous expressed sequence tags are candidates for contributing to interindividual blood pressure variation in the population at large by virtue of their positions within the region identified by linked marker loci. Our analysis also identified regions of the genome and candidate genes that are likely not to have a major impact on interindividual SBP variation in this population. Chromosome 8, for example, provided no evidence that it contains a gene that affect interindividual SBP variation. Each position on this chromosome had a P value between 0.57 and 1.0. As reported previously by other investigators using a candidate-gene approach, the renin gene on chromosome 1 did not yield any evidence for linkage to SBP Krushkal et al March 23, 1999 1409 Downloaded from http://circ.ahajournals.org/ by guest on August 11, 2017 Significant genome region identified by highly discordant sibling linkage design. Thin dotted lines indicate values of t statistic, whereas bold lines show significance levels for deviation of observed identity by descent sharing from that expected under the null hypothesis of no linkage (P values). Black triangles flank the regions with P,0.01. Location intervals for candidate genes are shown by bold horizontal lines on top of each panel. A, Chromosome 6 region. PLN indicates phospholamban; ESR, estrogen receptor. B, Chromosome 15 region. ANPEP indicates aminopeptidase N. C, Chromosome 5 region. ADRA1B indicates a1B-adrenergic receptor; DRD1A, dopamine receptor type 1A. D, Chromosome 2 region. CALM2 indicates calmodulin 2; NCX1, sodium-calcium exchanger 1. Candidate-gene locations were obtained from the Genome Database (http://www.gdbwww.gdb.org) and the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov). (P.0.13). Such negative results, however, should be interpreted with caution in light of the low power of genetic exclusion analyses for complex quantitative traits, such as blood pressure.11 We considered a probability value of ,0.01 in a region to be statistically significant. This is the same probability value suggested by others (eg, Rao12) for identifying regions for further investigation. We realize that this probability value for identification of regions of interest for positional candidate-gene analysis is greater than that suggested by Lander and Kruglyak.13 However, as discussed by many, such extreme probability values overemphasize the statistical interpretation or evaluation of a single study and will promote type II inferential errors (ie, failure to detect a blood pressure– controlling gene when it is really there). In addition, confident identification of linked regions, regardless of probability values, should not come from any single study but rather as the result of confirmation of linkage among similar populations of inference. Ultimately, however, the contributing genes must be identified and functional mutations characterized so that the underlying molecular and physiological mechanisms that relate gene variation to blood pressure variation can be unveiled. The results of this genome-wide linkage analysis of SBP provide a focus for future genetic studies. First, this study has identified chromosomal regions now hypothesized to contain blood pressure– controlling genes, which requires verification and additional testing. Second, it focuses the search for and cloning of new blood pressure–regulating genes. Finally, it provides a list of genes for detailed DNA sequence analyses aimed at identifying functional mutations that affect SBP variation. Identification of genes that contribute to blood pressure levels will provide a better understanding of the origin of high blood pressure and establish a basis for the development of improved prevention and treatment regimens. The data reported herein on the genetic contributors to interindividual SBP variation have implications not only for our understanding of high blood pressure but also for our understanding of the factors that contribute to its complications, such as cardiovascular, cerebrovascular, and renal diseases. Acknowledgments This study was supported by NIH grants HL-39107, HL-51021, HL-54481, HL-54457, and HL-54464 from the National Heart, Lung, and Blood Institute. 1410 Genome-Wide Analyses of Systolic Blood Pressure References 1. MacMahon S, Peto R, Cutler J, Collins R, Sorlie P, Neaton J, Abbott R, Godwin J, Dyer A, Stamler J. Blood pressure, stroke, and coronary heart disease, I: prolonged differences in blood pressure: prospective observational studies corrected for the regression dilution bias. Lancet. 1990;335: 765–774. 2. Whelton PK. Epidemiology of hypertension. Lancet. 1994;344:101–106. 3. Ward R. Familial aggregation and genetic epidemiology of hypertension. In: Laragh JH, Brenner BM, eds. Hypertension: Pathophysiology, Diagnosis, and Management. 2nd ed. New York, NY: Raven Press; 1995;1:67– 88. 4. Soubrier F. Blood pressure gene at the angiotensin I-converting enzyme locus: chronicle of a gene foretold. Circulation. 1998;97: 1763–1765. 5. Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, Gill JR Jr, Ulick S, Milora RV, Findling JW, Canessa CM, Rossier BC, Lifton RP. Liddle’s syndrome: heritable human hypertension caused by mutations in the b subunit of the epithelial sodium channel. Cell. 1994;79:407– 414. 6. Eaves L, Meyer J. Locating human quantitative trait loci: guidelines for the selection of sibling pairs for genotyping. Behav Genet. 1994;24: 443– 455. 7. Risch N, Zhang H. Extreme discordant sib pairs for mapping quantitative trait loci in humans. Science. 1995;268:1584 –1589. 8. Turner ST, Weidman WH, Michels VV, Reed TJ, Ormson CL, Fuller T, Sing CF. Distribution of sodium-lithium countertransport and blood pressure in Caucasians five to eighty-nine years of age. Hypertension. 1989;13:378 –391. 9. Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet. 1996;58:1347–1363. 10. Zini S, Fournie-Zaluski M-C, Chauvel E, Roques BP, Corvol P, LlorensCortes C. Identification of metabolic pathways of brain angiotensin II and III using specific aminopeptidase inhibitors: predominant role of angiotensin III in the control of vasopressin release. Proc Natl Acad Sci U S A. 1996;93:11968 –11973. 11. Page GP, Amos CI, Boerwinkle E. The quantitative LOD score: test statistic and sample size for exclusion and linkage of quantitative traits in human sibships. Am J Hum Genet. 1998;62:962–968. 12. Rao DC. CAT scans, PET scans, and genomic scans. Genet Epidemiol. 1998;15:1–18. 13. Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11: 241–247. Downloaded from http://circ.ahajournals.org/ by guest on August 11, 2017 Genome-Wide Linkage Analyses of Systolic Blood Pressure Using Highly Discordant Siblings Julia Krushkal, Robert Ferrell, Stephen C. Mockrin, Stephen T. Turner, Charles F. Sing and Eric Boerwinkle Downloaded from http://circ.ahajournals.org/ by guest on August 11, 2017 Circulation. 1999;99:1407-1410 doi: 10.1161/01.CIR.99.11.1407 Circulation is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231 Copyright © 1999 American Heart Association, Inc. All rights reserved. Print ISSN: 0009-7322. Online ISSN: 1524-4539 The online version of this article, along with updated information and services, is located on the World Wide Web at: http://circ.ahajournals.org/content/99/11/1407 Data Supplement (unedited) at: http://circ.ahajournals.org/content/suppl/1999/03/15/99.11.1407.DC1 Permissions: Requests for permissions to reproduce figures, tables, or portions of articles originally published in Circulation can be obtained via RightsLink, a service of the Copyright Clearance Center, not the Editorial Office. Once the online version of the published article for which permission is being requested is located, click Request Permissions in the middle column of the Web page under Services. Further information about this process is available in the Permissions and Rights Question and Answer document. Reprints: Information about reprints can be found online at: http://www.lww.com/reprints Subscriptions: Information about subscribing to Circulation is online at: http://circ.ahajournals.org//subscriptions/