Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Zinc finger nuclease wikipedia , lookup
Primary transcript wikipedia , lookup
Epigenomics wikipedia , lookup
Cancer epigenetics wikipedia , lookup
Gene therapy of the human retina wikipedia , lookup
Genome evolution wikipedia , lookup
Genetic engineering wikipedia , lookup
Microevolution wikipedia , lookup
Designer baby wikipedia , lookup
Extrachromosomal DNA wikipedia , lookup
Oncogenomics wikipedia , lookup
Molecular cloning wikipedia , lookup
Polycomb Group Proteins and Cancer wikipedia , lookup
Point mutation wikipedia , lookup
Mir-92 microRNA precursor family wikipedia , lookup
DNA vaccination wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Gel electrophoresis of nucleic acids wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Genomic library wikipedia , lookup
Cre-Lox recombination wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
History of genetic engineering wikipedia , lookup
Genome editing wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
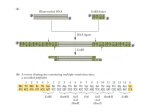
Construction and Validation of EBV-BACs Materials and Methods Generation of EBNA3 gene-deleted and revertant EBV and infection of BL cells The EBNA3 genes were deleted from the B95.8 EBV-BAC [p2089, (Delecluse et al., 1998)] by RecA-based homologous recombination. Regions of the EBV genome flanking EBNA3A, EBNA3B and EBNA3C were amplified by PCR and pairs of these products were cloned contiguously into the shuttle plasmid pKovKanΔCm [(White et al., 2003) and Supplementary Figure 1A]. Thus each shuttle plasmid contained a fragment of the EBV genome with an internal deletion of either EBNA3A [B95.8 (Acc#:V01555) coordinates 91915 to 95428 with internal deletion 92310 to 94900], EBNA3B (B95.8 coordinates 94901 to 98826 with internal deletion 95429 to 98233) or EBNA3C (B95.8 coordinates 98234 to 101482 with internal deletion 98827 to 100998). In the case of EBNA3C, only exon 2 (ex2) of the gene was deleted. The EBNA3 genes in the EBV-BAC were replaced by these deletion constructs by homologous recombination between the GFP-positive, hygromycin-resistant, B95.8 EBVBAC and these shuttle plasmids, as described previously [(White et al., 2003); Figure 1A and Supplementary Figure 1B]. Revertants were then made for each deletion by homologous recombination between the deletion BAC and a shuttle plasmid harbouring the appropriate EBNA3 gene, thus restoring the deleted gene. The integrity of the BACs was screened at each stage of recombination by miniprep DNA isolation, restriction digest and pulsed field gel electrophoresis. 293 cell clones containing deletion and revertant BACs were produced: BAC DNA was purified by maxiprep (Qiagen) and 1 µg transfected into 293 cells using an integrin-targeting peptide combined with Lipofectin (Invitrogen) as described previously (Hart et al., 1998). Cells were exposed to lipid-peptideDNA complexes for 4 to 6 hours in Optimem (Invitrogen). Stable 293 cell clones were isolated under hygromycin B selection (Roche; 125 µg/ml) by ring and dilution cloning. A 293 clone containing the BAC used to generate the recombinants (293-2089-F2) was also produced. Episomal BAC DNA was recovered from 293 cells grown in six-well dishes by preparation of low molecular weight DNA as described previously (Wade-Martins et al., 1999) and 4% electroporated into Electromax™ DH10B™ E.coli (Invitrogen). Several individual colonies were selected and DNA extracted by miniprep and analysed by restriction digest and pulsed field gel electrophoresis. Infectious virus was generated from 293 producer cells by transient transfection of BZLF1 and BALF4 expression plasmids (Dirmeier et al., 2003; Neuhierl et al., 2002). Supernatant was harvested and infectivity assessed by GFP expression in Raji cells. One ml of supernatant containing EBNA3A, EBNA3B and EBNA3C knockout and revertant viruses, and the wild type virus (F2), was used to infect 1x105 EBV-negative BL cells (BL31 and BL2). After 48 hours, cells were selected in Hygromycin B (Roche) at a concentration of 200 µg/ml to produce EBV-converted BL lines. Episomal BAC DNA was recovered from 1x106 EBV-converted BL cells by the same method as described for 293 cells. Integrity of the BAC was assessed by restriction digest and pulsed field gel electrophoresis. Results Screening of recombinant EBV-BACs Individual EBNA3 genes were deleted from the EBV-BAC by homologous recombination with an appropriate shuttle plasmid (Figure 1A and Supplementary Figure 1). In order to control for any second site mutations that may have occurred, each gene was replaced during a second round of recombination to generate revertant BACs. Deletion of each EBNA3 gene introduced either a Cla I or Not I restriction site into the EBV-BAC (the location of these restriction sites in the original EBV-BAC is shown in Supplementary Figure 1A). Digestion with these restriction enzymes followed by pulsed field gel electrophoresis therefore enabled diagnosis of deletion and revertant BACs, with the size of restriction fragments from different deletion mutants and the original EBV-BAC being sufficiently different to allow discrimination by pulsed field gel electrophoresis alone (Supplementary Figures 2A and 2B). Two different EBNA3C-knockout BACs were generated; one in which only the expected change (deletion of EBNA3C) had taken place (3CKO), and one which had also gained DNA in the Bam W repeat region (3CKO+BamW). Pulsed field gel analysis of 3CKO+BamW digested with various different restriction enzymes suggested that expansion of this region corresponds to addition of two copies of the Bam W repeat and was the only gross rearrangement that had occurred (data not shown). Given the considerable variation in the number of Bam W repeats in different EBV strains and the fact that the repeat number found in 3CKO+BamW was well within this range, expansion of this region was not considered to be undesirable. Wild type, deletion and revertant BACs were transfected into 293 cells and stable producer cell clones generated. The integrity of episomal BAC DNA recovered from 293 producer cells was analysed by restriction digest with Eco RI (Supplementary Figure 3) and Age I (data not shown) and pulsed field gel electrophoresis. These restriction enzymes cut throughout the EBV genome allowing any gross rearrangements to be easily identified by comparison with the BAC DNA originally transfected. Validation of recombinant viruses in BL31 cells Infectious virus was generated from wild type, deletion and revertant BACs maintained in 293 cells and used to infect EBV-negative BL31 cells. Viruspositive converts were selected in hygromycin and analysed for EBV gene expression (shown in Figure 1C and discussed in results section). Episomal BAC DNA was also recovered from converted BL31 cell lines and its integrity assessed by restriction digest with Eco RI and pulsed field gel electrophoresis (data not shown). BACs rescued from many of the BL31 lines had undergone some alteration, but the vast majority of these changes were confined to either the Bam W repeat region or the terminal repeat region. It was assumed that loss or gain of repeat elements in these regions, within certain limits, would not affect the function of the EBV episome within BL31 cells, as verified by the correct expression of EBV latent genes (Figure 1C).