Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Plant nutrition wikipedia , lookup
Microbial metabolism wikipedia , lookup
Adenosine triphosphate wikipedia , lookup
Photosynthesis wikipedia , lookup
Mitochondrion wikipedia , lookup
Butyric acid wikipedia , lookup
Basal metabolic rate wikipedia , lookup
Biosynthesis wikipedia , lookup
Lactate dehydrogenase wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Fatty acid synthesis wikipedia , lookup
Biochemistry wikipedia , lookup
Fatty acid metabolism wikipedia , lookup
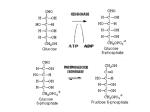
Stephanie Hickey Nutrition 445 Shearer 5 March 2014 Case Study 2 Pyruvate carboxylase deficiency & dysfunction of the Krebs cycle: A 3-month old female infant seemed normal until she developed seizures. The infant became progressively worse, showing hypotonia, psychomotor retardation, and poor head control. She had lactic acidosis and an elevated plasma pyruvate level, both more than seven times the normal amount. Plasma alanine concentration was high, and an alanine load failed to induce a normal gluconeogenic response. Pyruvate carboxylase activity was measured using extracts of cultured skin fibroblasts and was found to be less than 1% of the normal level. Both the mother and the father had intermediate levels of fibroblastic pyruvate carboxylase. Fibroblasts from the patient accumulated five times greater than normal amounts of lipid. Questions: 1. What reaction is catalyzed by pyruvate carboxylase? And where is the enzyme located within cells? Pyruvate carboxylase, a mitochondrial enzyme regulated by acetyl-CoA, is crucial for intermediary metabolism, controlling fuel portioning toward gluconeogenesis, lipogenesis, biosynthesis of neurotransmitter substances and glucose-induced insulin secretion by pancreatic islets. It catalyzes the HCO3 and MgATP-dependent carboxylation of pyruvate to form oxaloacetate. This conversion of pyruvate to oxaloacetate by pyruvate carboxylase is called an anaplerotic process because of its role in restoring oxaloacetate to the TCA cycle (Gropper & Smith, 2013). In other words, pyruvate carboxylase supplies oxaloacetate to keep the TCA cycle running. For example: (Precursors for gluconeogenesis , 2013) Hickey 2 Pyruvate carboxylase is found in both prokaryotes and eukaryotes including fungi, bacteria, plants and animals. It occurs in two distinct isoforms, one in the mitochondria and the other in the cytoplasm. 2. What is the metabolic function of pyruvate carboxylase? Pyruvate carboxylase plays a role in gluconeogenesis, which is the formation of glucose by the liver or kidney from noncarbohydrate sources. Pyruvate carboxylase catalyzes the first step in gluconeogenesis, converting pyruvate to oxaloacetate for conversion into phosphoenolpyruvate (PEP) by phosphoenolpyruvate carboxykinase (PEPCK) which gets transported to the TCA cycle where is gets converted to glucose. Pyruvate carboxylase ensures that there is a constant supply of oxaloacetate for the TCA cycle by forming oxaloacetate directly from pyruvate by the addition of carbon, this addition of carbon dioxide happens due to ATP and biotin. Pyruvate Carboxylase also plays a role in adipose tissues because it provides oxaloacetate to assist in the export of acetyl-CoA during the formation citrate in the mitochondria for fatty acid synthesis. Fatty acid synthase catalyzes the action of two-carbon molecules from malonyl-CoA to produce a long chain fatty acid that are esterified with glycerol to form triacylglycerol (Jitrapakdee, et al., 2008). Pyruvate carboxylase can alter the rate of glutamate synthesis by increasing the amount of TCA cycle intermediates. The role of pyruvate carboxylase in astrocytes is that it can utilize α-ketogluterate as a precursor for the formation of glutamate. Since glutamate is constantly being withdrawn from the TCA cycle, pyruvate carboxylase is responsible to replenish this intermediate in the astrocyte. 3. Explain the failure of the alanine load to induce gluconeogenesis in the patient? Alanine is the main amino acid extracted from the skeletal muscle in both post absorptive state and after several weeks of starvation. The decline in the rate of gluconeogenesis results from a reduction of alanine supply to the liver. Failure of alanine supply leads to a failure of pyruvate supply that prevents the TCA cycle from happening and therefore prevents oxaloacetate converting to malate and then malate can’t leave the mitochondria and can’t induce gluconeogenesis in the cytosol. The glucose-alanine cycle, which is alanine coming from the skeletal muscle pyruvate in mitochondria oxaloacetate in the TCA cycle malate in the TCA cycle malate in the cytosol oxaloacetate gluconeogenesis, is stopped which is pyruvate generated within the skeletal muscle can be transaminated by ALT to alanine which is returned back to the liver from the muscle for gluconeogenesis. The alanine is then converted to pyruvate by the transfer of the amino group to an α-ketogluterate in the conversion of alanine in the liver. This involves the utilization of lactate, produced by glycolysis in the muscle as a carbon source for hepatic gluconeogenesis. So the liver can convert lactate back into glucose for reuse by the muscle Hickey 3 (Gropper & Smith, 2013). The amino group transported from the muscle to the liver in the form of alanine is converted to urea in the urea cycle and excreted (Felig, Pozefsky, Marliss, & Cahill, 1970). Image: (Chhabra, 2012) This cycle provides a means for localized ATP production and a way to convert muscle glycogen into blood glucose. During failure of alanine load to induce gluconeogenesis results in an overall net loss of ATP and excess of lactic acid within the body. 4. Glutamine greatly simulated the growth of fibroblasts from a patient with pyruvate carboxylase deficiency. Why? In normal individuals, oxaloacetate may be produced by pyruvate through the intermediate of pyruvate carboxylase, but in those with a pyruvate carboxylase deficiency this reaction would be decreased. Pyruvate carboxylase deficiency is a rare genetic disorder that affects infants who develop severe lactic acidosis within the first few days of birth. Supplementation of glutamine, glutamic acid, aspartic acid and asparagine will increase survival rate. These amino acids provide oxaloacetate in the TCA cycle for oxidative metabolism in making ATP. Glutamine is relied on for the maintenance of growth of skin fibroblast cells compared to normal cells. In normal individuals without the deficiency, cultured skin fibroblasts can readily oxidize leucine to carbon dioxide by the acetyl-CoA and the TCA cycle intermediates. Individuals with a pyruvate carboxylase deficiency produce less carbon dioxide from leucine than those without the deficiency due to the fact that interference with intermediates results in low production of carbon dioxide. The requirement of glutamine is greater in those with pyruvate carboxylase deficiency than Hickey 4 those without the deficiency because the defective cells have a shorter life span when cultured with glutamine. This can explain the dysfunction of the TCA cycle due to not enough oxaloacetate being generated; glutamine can provide a source of oxaloacetate by forming 2-oxogluterate (Oizumi, Ng, & Donnell, 1986) . 5. What treatment would you suggest for a patient with this disease? Treatment of a deficiency in pyruvate carboxylase includes providing alternative energy sources and means of metabolizing pyruvate for the body. I would suggest supplementing the individual with biotin, citrate, and aspartic acid. Biotin has the ability to improve the function of the pyruvate carboxylase enzyme activity (Ahmad, et al., 1999). Citrate can help reduce the level of lactic acid and provides the substrate that is needed in the TCA cycle (Ahmad, et al., 1999). Aspartic acid supplementation allows the urea cycle to proceed by reducing the ammonia levels (Wang & De, 2011). Hickey 5 Works Cited Ahmad, Ayesha, Stephen G. Kahler, Priya S. Kishnani, Merce Artigas-Lopez, Anuradha S. Pappu, Robert Steiner, David S. Millington, and Johan L.K. Van Hove. "Treatment of Pyruvate Carboxylase Deficiency With High Doses of Citrate and Aspartate." American Journal of Medical Genetics 87.4 (1999): 331-38. PubMed. Web. 10 Oct. 2013. Chhabra, N. (2012, July 19). Gluconeogenesis - Subjective questions. Retrieved from Biochemistry for Medics: http://www.namrata.co/gluconeogenesis-subjective-questionssolved-part-1/ Felig, Philip, Thomas Pozefsky, Errol Marliss, and George F. Cahill, Jr. "Alanine: Key Role in Gluconeogenesis." American Association for the Advancement of Science 3920th ser. 167 (1970): 1003-004. JSTOR. Web. 9 Oct. 2013. Oizumi, J., W. G. Ng, and G.N. Donnell. "Pyruvate Carboxylase Defect: Metabolic Studies on Cultured Skin Fibroblasts." J. Inher. Metab. Dis. 9 (1986): 120-28. PubMed. Web. 10 Oct. 2013. Precursors for gluconeogenesis . (2013, October 7). Retrieved from ExpertsMind.com: http://www.expertsmind.com/topic/gluconeogenesis/precursors-for-gluconeogenesis95895.aspx Smith, Jack L. "3, 7, 9." Advanced Nutrition and Human Metabolism. By Sareen S. Gropper. Sixth ed. Belmont: Wadsworth Cengage Learning, 2013. 89, 259, 341+. Print. Wang, Dong, and Darryl De Vivo. "Pyruvate Carboxylase Deficiency." NCBI. GeneReview, 21 July 2011. Web. 10 Oct. 2013.