Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Artificial gene synthesis wikipedia , lookup
Molecular evolution wikipedia , lookup
Protein (nutrient) wikipedia , lookup
Bottromycin wikipedia , lookup
List of types of proteins wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Protein moonlighting wikipedia , lookup
Gene expression wikipedia , lookup
Multi-state modeling of biomolecules wikipedia , lookup
Protein adsorption wikipedia , lookup
Western blot wikipedia , lookup
Protein design wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
Drug design wikipedia , lookup
Rosetta@home wikipedia , lookup
Protein–protein interaction wikipedia , lookup
Intrinsically disordered proteins wikipedia , lookup
Nuclear magnetic resonance spectroscopy of proteins wikipedia , lookup
Protein structure prediction wikipedia , lookup
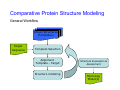
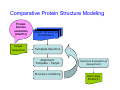
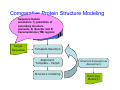
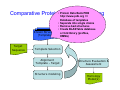
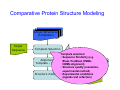
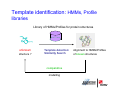
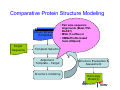
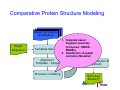
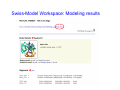
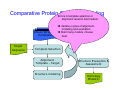
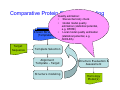
Introduction to 3D-Structure Visualization and Homology Modeling using the Swiss-Model Workspace Lorenza Bordoli Biozentrum of the University of Basel and Swiss Institute of Bioinformatics Lecture 3 May 2009 Lecture 3: Outline • Homology modeling using the SwissModel Workspace: – Target sequence feature annotation – Template identification – Target-template alignment – Homology Modeling by template based fragment assembly – Model quality estimation Homology (Comparative) Modeling Evolution of Protein Structures Protein structure is better conserved than sequence Similar Sequence Î Similar Structure Homology modeling = Comparative protein modeling Idea: Using experimental 3D-structures of related family members (templates) to calculate a model for a new sequence (target). Comparative Protein Structure Modeling General Workflow Known Structures (Templates) Target Sequence Template Selection Alignment Template - Target Structure Evaluation & Assessment Structure modeling Homology Model(s) Protein structure homology modeling using SWISS-MODEL workspace • Freely accessible: http://swissmodel.expasy.org/workspace Comparative Protein Structure Modeling Protein Domain annotation (InterPro) Target Sequence Known Structures (Templates) Template Selection Alignment Template - Target Structure Evaluation & Assessment Structure modeling Homology Model(s) Functional annotation of multi-domain proteins MSA MSA Model (HMM, PSSM,…) for Model for Model for DNA bdg. Function Activation Function 1 MSA Activation Function 2 InterPro http://www.ebi.ac.uk/interpro/ InterPro is a database protein families, domains, regions, repeats and sites in which identifiable features found in known proteins can be applied to new protein sequences. Member Databases: – – – – – – – – – Pfam (HMMs) PRINTS (PSSMs) PROSITE (Patterns & Profiles) ProDom (Motifs from PsiBlast) SMART (HMMs) TIGRFAMs (HMMs) PANTHER (HMMs) Superfamily (HMMs from SCOP) UniProt (Sequences) InterPro 1. Family: An InterPro family is a group of evolutionarily related proteins that share similar domain (or repeat) architecture. 2. Domain: An InterPro domain is an independent structural unit, which can be found alone or in conjunction with other domains or repeats. Domains are evolutionarily related. 3. Repeat: An InterPro repeat is a region that is not expected to fold into a globular domain on its own. For example 6-8 copies of the WD40 repeat are needed to form a single globular domain. 4. PTM site: A post-translational modification modifies the primary protein structure. This modification may be necessary for activation or de-activation of function. Examples include glycosylation, phosphorylation, and sulphation, splicing etc. 5. Binding site: An InterPro Binding site binds chemical compounds, which themselves are not substrates for a reaction. The compound, which is bound, may be a required co-factor for a chemical reaction, be involved in electron transport or be involved in protein structure modification. 6. Active site: Active sites are best known as the catalytic pockets of enzymes where a substrate is bound and converted to a product, which is then released. Distant parts of a protein's primary structure may be involved in the formation of the catalytic pocket. Therefore, to describe an active site, different signatures will be needed to cover the active site residues. Functional and structural domain annotation • Individual structural domains of multidomain proteins often correspond to units of distinct molecular function => you can compare the functional domain prediction with the functional annotations of detected templates. • The sensitivity of profile-based template detection methods can be enhanced when the search is performed at the domain level rather than searching the whole protein sequence. Comparative Protein Structure Modeling Sequence feature annotation: 1) prediction of secondary structure elements, 2) disorder and 3) Known transmembrane (TM)Structures regions (Templates) Target Sequence Template Selection Alignment Template - Target Structure Evaluation & Assessment Structure modeling Homology Model(s) Target sequence feature annotation • In the twilight zone of sequence alignments, applying secondary structure prediction to the protein of interest may help deciding whether a putative template shares essential structural features. • The sensitivity of profile-based template detection methods can be enhanced when the search is performed at the domain level rather than searching the whole protein sequence. Target sequence feature annotation • Intrinsically unstructured (disordered) regions in proteins have been associated with numerous important biological cellular functions (e.g. cell signaling, transcriptional regulation,…) • Prediction of disordered and transmembrane regions therefore complement the analysis of protein domain boundaries and functional annotation of the target protein. Swiss-Model Workspace: Sequence feature annotations Swiss-Model Workspace: Sequence feature annotations Human Rhodopsin Protein Data BankModeling PDB Comparative Protein• Structure http://www.pdb.org => Known Structures (Templates) Target Sequence • • • Database of templates Separate into single chains Remove bad structures Create BLASTable database or fold library (profiles, HMMs) Template Selection Alignment Template - Target Structure Evaluation & Assessment Structure modeling Homology Model(s) Swiss-Model Workspace: Template Library Swiss-Model Workspace: Template Library Comparative Protein Structure Modeling Known Structures (Templates) Target Sequence Template Selection Template selection: • Sequence Similarity (e.g. Alignment Blast, Psi-Blast, HMMs-Evaluation & Structure Template - Target HMMs alignment) Assessment • Structure quality (resolution, experimental method) Structure modeling • Experimental conditions Homology (ligands and cofactors) Model(s) Template identification: Blast, PSI-Blast PDB: database of protein structures unknown structure ? Template detection: Similarity Search comparative modeling Alignment to similar protein with known structure Template identification using fold libraries Example : Leucin Zipper DNA binding Domain of Transcription Factors. Human c-fos/c-jun Structural superposition Yeast bZIP MSA … Model (HMM, PSSM,…) for Structural domains (e.g. TF DNA binding domain) => Library of folds Structural Alignments • Protein structure is better conserved than sequence • Structural alignments establish equivalences between amino acid residues based on the 3D structures of two or more proteins. • Structure alignments therefore provide information not available from sequence alignment methods alone. Template identification: HMMs, Profile libraries Library of HMMs/Profiles for protein structures unknown structure ? Template detection: Similarity Search comparative modelling Alignment to HMMs/Profiles of known structures Swiss-Model Workspace: Template identification Swiss-Model Workspace: Template identification Swiss-Model Workspace: Template identification Swiss-Model Workspace: Template identification Swiss-Model Workspace: DeepView Project file Comparative Protein Structure Modeling • Pair wise sequence alignments (Blast, PSIBLAST) Known Structures • MSA (T-coffee) or (Templates) • HMMs/Profile based tools (HHpred) Target Sequence Template Selection Alignment Template - Target Structure Evaluation & Assessment Structure modeling Homology Model(s) Swiss-Model Workspace: target-template alignment • The target–template sequence alignments generated by the different template database search techniques can be used as the basis for the subsequent model creation. The alignments can be downloaded as DeepView project file, which contains the target sequence aligned to the template structure. • The program DeepView allows you to display and analyze the alignment in the structural context of the template to manually adjust misaligned regions. • Once you have finished editing the alignment, save the project file on the local disk and submit it to the ‘Project Mode’ of the Modeling session for model building DeepView: “Manual Modeling” [ http://www.expasy.org/spdbv/ ] Swiss-Model Workspace: Modeling Swiss-Model Workspace: target-template alignment • You might also want to apply alternative sequence alignment methods (see Lecture 2) by using multiple sequence alignment programs to align the target and the template sequences and family related proteins. • The obtained sequence alignment between target and template (and additional homologous proteins) can be submitted to the ‘Alignment mode’ of the modeling session for model building. Swiss-Model Workspace: Modeling Comparative Protein Structure Modeling Known Structures (Templates) Target Sequence 1. Template based fragment assembly (Composer, SWISSTemplate Selection MODEL) 2. Satisfaction of spatial restraints (Modeller) Alignment Structure Evaluation & Template - Target Assessment Structure modeling Homology Model(s) Swiss-Model: Template based fragment assembly • Find structurally conserved core regions Swiss-Model: Template based fragment assembly • Build model core – … by averaging core template backbone atoms (weighted by local sequence similarity with the target sequence). Leave non-conserved regions (loops) for later … Swiss-Model: Template based fragment assembly • Loop (insertion) modeling – Use the “spare part” algorithm to find compatible fragments in a Loop-Database, or “ab-initio” rebuilding (e.g. Monte Carlo, MD, GA, etc.) to build missing loops. Swiss-Model: Template based fragment assembly • Side Chain placement – Find the most probable side chain conformation, using • homologues structure information • back-bone dependent rotamer libraries • energetic and packing criteria Swiss-Model: Template based fragment assembly • Rotamer Libraries – Only a small fraction of all possible side chain conformations is observed in experimental structures – Rotamer libraries provide an ensemble of likely conformations – The propensity of rotamers depends on the backbone geometry: Swiss-Model Workspace: Modeling results Swiss-Model Workspace: Modeling results Comparative ProteinErrors Structure Modeling in template selection or alignment result in bad models: Î iterative cycles of alignment, modeling and evaluation Known Structures Î Built many models, choose (Templates) best. Target Sequence Template Selection Alignment Template - Target Structure Evaluation & Assessment Structure modeling Homology Model(s) Comparative ProteinQuality Structure Modeling estimation: • • Known Structures • (Templates) Target Sequence Stereochemistry check Global model quality estimation (statistical potential, e.g. DFIRE) Local model quality estimation (statistical potential, e.g. ANOLEA) Template Selection Alignment Template - Target Structure Evaluation & Assessment Structure modeling Homology Model(s) Model quality estimation • Ramachandran Plot of backbone angles (ϕ,ψ) – favored regions – generously allowed regions – disallowed regions – Amino acids with special properties: • PRO: ϕ = 60º • GLY ( ) • Similar plots for χ-angle distributions Î Useful to identify regions with errors in geometry ANOLEA : (Atomic Non-Local Environment Assessment) • http://protein.bio.puc.cl/cardex/servers/anolea/ • http://swissmodel.expasy.org/anolea/ ANOLEA Correct Structure: PDB: 1GES Î Detects local packing errors Î Errors in alignments Model with wrong alignment: Swiss-Model Workspace: Quality estimation All checking tools are happy, so can I believe it now? Models are not experimental facts ! Models can be partially inaccurate or sometimes completely wrong ! A model is a tool that helps to interpret biochemical data. References and further reading: References and further reading: