Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Vasopressin wikipedia , lookup
Hypothalamus wikipedia , lookup
Hormone replacement therapy (male-to-female) wikipedia , lookup
Hormone replacement therapy (female-to-male) wikipedia , lookup
Signs and symptoms of Graves' disease wikipedia , lookup
Hypoglycemia wikipedia , lookup
Androgen insensitivity syndrome wikipedia , lookup
Hypothyroidism wikipedia , lookup
Graves' disease wikipedia , lookup
Hyperthyroidism wikipedia , lookup
Growth hormone therapy wikipedia , lookup
Hyperandrogenism wikipedia , lookup
Congenital adrenal hyperplasia due to 21-hydroxylase deficiency wikipedia , lookup
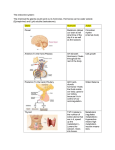
CM E Endocrine Disorders in the Neonatal Period Karishma A. Datye, MD; and Andrew A. Bremer, MD, PhD CM E Abstract Many endocrine conditions are unique to the perinatal period. In this article, we review many such conditions, including disorders of the pituitary gland, disorders of sexual differentiation, disorders of glucose homeostasis, disorders of the thyroid gland, and disorders of calcium homeostasis. Rather than serving as a comprehensive resource, the article is meant to serve as a guide for general pediatricians and neonatologists caring for infants with endocrine disorders. Moreover, because the field of pediatric endocrinology continues to evolve, consultation with a pediatric endocrinologist for any child with an endocrinopathy is recommended. EDUCATIONAL OBJECTIVES 1. Review common endocrine conditions that often present in the newborn and perinatal period. 2. Determine the enzymes involved in steroidogenesis. 3. Discuss the diagnostic evaluation of hypoglycemia. Karishma A. Datye, MD, is a PGY-3 Resident in Pediatrics, Vanderbilt University School of Medicine. Andrew. A. Bremer, MD, PhD, is Assistant Professor of Pediatrics, Division of Pediatric Endocrinology, Department of Pediatrics, Vanderbilt University © Shutterstock School of Medicine. Address correspondence to: Andrew A. Bremer, MD, PhD, Department of Pediatrics, Division of Endocrinology, Vanderbilt University, 2200 Children’s Way, 11134-A DOT-9170, Nashville, TN 37232-9170; email: [email protected]. Disclosure: The authors have no relevant financial relationships to disclose. doi: 10.3928/00904481-20130426-08 PEDIATRIC ANNALS 42:5 | MAY 2013 Healio.com/Pediatrics | 67 CM E T he transition from the intrauterine to extrauterine environment is a “rite of passage” for every individual. However, the perinatal period is also a time when an infant may display clinical signs of an underlying endocrinopathy that may not have been apparent either in utero or at birth. Herein, we present some common endocrinopathies that may present in the neonatal period. Importantly, the descriptions are not intended to replace the more comprehensive information that can be found in general pediatric or endocrine texts; rather, they are meant to serve as a quick guide for general pediatricians and neonatologists caring for infants with endocrine disorders. As with most endocrine conditions in the perinatal period, consultation with a pediatric endocrinologist is recommended regarding the most up-to-date treatment. DISORDERS OF THE PITUITARY GLAND The pituitary gland is critical to human development, and secretes many hormones vital to normal growth and survival. Importantly, the pituitary gland is divided into two distinct parts; the adenohypophysis (composed of the anterior lobe of the pituitary, intermediate lobe, and pars tuberalis) is derived from the oral ectoderm,1 whereas the neurohypophysis (or posterior pituitary gland) is derived from the neural ectoderm of the forebrain.2 The blood supply to these embryologically distinct parts of the pituitary also differ, with that of the adenohypophysis coming from the hypothalamic-pituitary portal circulation and that of the neurohypophysis coming from the inferior hypophyseal artery.1 The adenohypophysis, after receiving myriad signaling factors from the hypothalamus through the hypothalamic-pituitary portal system, produces several hormones necessary for growth and development. Five different cell types in the anterior pituitary secrete six 68 | Healio.com/Pediatrics separate hormones: follicle-stimulating hormone (FSH), leutinizing hormone (LH), thyroid-stimulating hormone (TSH), growth hormone (GH), prolactin (PRL), and adrenocorticotropic hormone (ACTH).2 Alternatively, the posterior pituitary releases hormones important in the regulation of blood volume (arginine vasopressin [AVP]) and reproduction (oxytocin).2 The nonspecific signs and symptoms of hypopituitarism in the neonatal period can make its diagnosis difficult. Hypopituitarism in the neonatal period can be secondary to genetic or anatomic abnormalities, and can present as isolated or multiple hormone deficiencies. One genetic cause of combined pituitary hormone deficiencies is mutations in the PROP1 gene, which regulates the development of several anterior pituitary cell lineages.2 Anatomic abnormalities that may cause pituitary dysfunction include midline defects like septo-optic dysplasia and optic nerve hypoplasia.2 The nonspecific signs and symptoms of hypopituitarism in the neonatal period can make its diagnosis difficult. Infants may present with poor growth, poor feeding, temperature instability, or hypoglycemia.2 In males, the combination of micropenis and hypoglycemia should always alert the clinician of potential pituitary dysfunction. Early diagnosis is important as prolonged hypoglycemia, hypothyroidism, in addition to other manifestations of hypopituitarism, have the potential to cause developmental delays. Central adrenal insufficiency could also potentially be life-threatening. Hypopituitarism is diagnosed through a combination of individual hormone testing (eg, evaluating the thyroid axis, ad- renal axis, growth hormone axis, etc), in addition to magnetic resonance imaging (MRI) of the pituitary gland and hypothalamus.2 Treatment varies depending on the specific hormones affected. The posterior pituitary gland releases AVP as a mechanism to regulate the volume status of the body. AVP then acts on the kidney to control the amount of water that is excreted from the body.3 Central diabetes insipidus (DI) occurs when defects in vasopressin at the level of the pituitary lead to inappropriate water loss and large quantities of dilute urine.4 The etiology of central DI varies by age group; in the neonatal period, the majority of central DI is caused by anatomic or genetic defects. Examples of anatomic abnormalities include septooptic dysplasia, agenesis of the corpus callosum, and holoprosencephaly.4 Both autosomal dominant and recessive genetic defects have also been identified that cause central DI.5 Other causes of central DI include acquired DI after surgery, for example after craniopharyngioma resection, or direct invasion of the posterior pituitary by tumor.4 Infants with DI may present with polyuria, polydipsia, and poor growth, in addition to other non-specific symptoms. Biochemical abnormalities include serum hyperosmolality (typically with concomitant hypernatremia) in the setting of dilute urine. In older patients, the diagnosis may be made through water deprivation testing; however, in neonates, making the diagnosis can be challenging because water deprivation cannot be easily performed. If central DI is suspected, an MRI of the pituitary should be performed to evaluate for midline structural defects6 and the posterior pituitary “bright spot” (signifying the anatomic presence of AVP). Central DI is typically managed by administering desmopressin, a vasopressin analog.6 In neonates, administration of this analog coupled with polydipsia may lead to hyponatremia; therefore, in some cases, PEDIATRIC ANNALS 42:5 | MAY 2013 CM E fluid intake (to match urinary losses) is the sole treatment.6 Unlike central DI, nephrogenic DI occurs when the kidney is resistant to the actions of vasopressin. Nephrogenic DI can result from genetic or acquired causes, and since the kidney is typically nonresponsive to vasopressin, desmopressin in most cases is not a therapeutic option. Thus, the main goals of therapy are to ensure the intake of adequate calories for growth and to avoid severe dehydration. Thiazide diuretics in combination with amiloride or indomethacin are also often used. DISORDERS OF SEXUAL DIFFERENTIATION Normal sexual development consists of three related and sequential processes: 1) the establishment of the chromosomal (genetic) sex (XX as female, XY as male); 2) the determination of the gonadal sex; and 3) the development of the sexual phenotypes. For the first 2 months after fertilization, the 2 sexes develop in an identical fashion. Then, around the 8th week of gestation, the indifferent gonad develops into an ovary or testis, with subsequent hormone production in the case of testicular development. The hormonal profile then directs the development of the indifferent anlagen of the urogenital tract into characteristic male or female structures, a process which is largely completed by the 12th week of gestation. Although an extensive review of all the disorders of sexual development (DSDs) is beyond the scope of this article, DSDs are often broken down into 3 categories: overvirilized 46,XX females, undervirilized 46,XY males, and ovotesticular DSDs. Overvirilized 46,XX Infants Overvirilization of 46,XX fetuses can occur by three main mechanisms: 1) increased androgen production, 2) decreased androgen metabolism (ie, clearance), and 3) maternal androgen expo- PEDIATRIC ANNALS 42:5 | MAY 2013 Figure 1. Integrated view of steroidogenesis. ACTH = adrenocorticotropic hormone; b5 = cytochrome b5; CMO = corticosterone methyloxidase; DHEA = dehydroepiandrosterone; DHT = dihydrotestosterone; DOC = deoxycorticosterone; HSD = hydroxysteroid dehydrogenase; POR = P450 oxidoreductase; StAR = steroidogenic acute regulatory protein. (Adapted from Oberfield et al29) sure (either endogenous or exogenous). The most common cause of overvirilized 46,XX fetuses is congenital adrenal hyperplasia (CAH), which is typically due to defects in the gene encoding the 21-hydroxylase enzyme (P450c21). In this condition, defects in 21-hydroxylase (and less commonly 11beta-hydroxylase [encoded by P450c11b]) lead to decreased cortisol synthesis (see Figure 1),7 which causes oversecretion of ACTH from the pituitary and ultimately increased androgen production and virilization of the external genitalia. A deficiency in 21-hydroxylase can also cause potentially life-threatening salt wasting and adrenal insufficiency, while 11betahydroxylase deficiency can cause fluid retention and hypertension secondary to deoxycorticosterone (DOC) production. Importantly, the internal genitalia (Mullerian structures) of these patients develop appropriately.5 Diagnosis of these disorders is based on build up of 17-hydroxyprogesterone in 21-hydroxylase deficiency7 and DOC and 11-deoxycortisol in 11beta-hydroxylase deficien- cy.5 Treatment includes prompt initiation of glucocorticoid therapy (with possible mineralocorticoid therapy as well) and referral to a pediatric endocrinologist. Another cause of virilization of the 46,XX fetus is decreased androgen metabolism, an example being placentalfetal aromatase deficiency. Normally, the testosterone that is derived from both the fetal and maternal production of dehydroepiandrosterone (DHEA) is converted to estradiol by the aromatase enzyme, “protecting” the female fetus from excessive androgen exposure. However, a deficiency in aromatase increases circulating testosterone levels and can cause virilization in both the mother and fetus.8 A further cause of virilization of the 46,XX fetus is increased maternal androgen exposure. Both endogenous (androgen-secreting adrenal or ovarian tumors)9 or exogenous androgens can cause virilization.10 However, since exposure to these androgens ceases after delivery, virilization of the infant will not continue to progress after birth. Healio.com/Pediatrics | 69 CM E Undervirilized 46,XY Infants Undervirilization of 46,XY fetuses, formerly called male pseudohermaphroditism, is caused by myriad factors resulting in errors of testosterone production, testosterone metabolism, and/ or end-organ action of testosterone. As early as 10 weeks gestation, testosterone is released from the Leydig cells in the testes to help form (along with dihydrotestosterone [DHT]) the male reproductive tissues and external genitalia. Early during gestation, human chorionic gonadotropin (hCG) from the placenta is responsible for testosterone secretion; later, LH from the developing fetal pituitary takes over. Both LH and hCG bind to the same receptor, and thus defects in this receptor can ultimately cause problems with testosterone secretion and abnormalities in external genitalia formation.11 As would be expected, defects in any enzyme associated with testosterone biosynthesis (see Figure 1) will result in undervirilization of 46,XY fetuses. Moreover, defects in gonadal and adrenal steroidogenesis (which occurs in congenital lipoid adrenal hyperplasia and is characterized by the impaired ability to convert cholesterol to pregnenolone12) also result in undervirilization of the 46,XY infant. A deficiency in 17alphahydroxylase/17,20-lyase also yields poor production of cortisol and testosterone (causing cortisol deficiency and undervirilization), as well as overstimulation of the mineralocorticoid pathway due to excess ACTH release from the pituitary (causing hypertension). Diagnosis of these conditions involves identifying elevated precursors in the steroiodogenesis pathway, and therapy involves treating salt wasting and cortisol deficiency (if present). Other causes of undervirilization of 46,XY fetuses include a deficiency in 5alpha-reductase, the enzyme that converts testosterone to the more potent androgen, DHT,13 as well as defects in 70 | Healio.com/Pediatrics the androgen receptor (AR) gene, which causes androgen insensitivity syndrome (AIS). Infants who are 46,XY with the complete form of AIS have female external genitalia and typically do not have Wolffian or Mullerian structures. Par- There is considerable debate regarding what glucose concentrations define hypoglycemia in the neonate. tial AIS in 46,XY infants causes variable degrees of undervirilization, and patients may have ambiguous external genitalia with Wolffian structures internally.14 Diagnosis is made by sequencing the AR gene, and by examining LH and testosterone levels. In complete AIS, sex assignment is female, whereas sex assignment in incomplete AIS is more challenging.5 Ovotesticular DSD Ovotesticular DSD, previously called true hermaphroditism, refers to individuals with ovarian and testicular tissues. The majority of these patients are 46,XX, although 46,XY and 45,X/46,XY patients are also seen.5 Gonads in these patients may be ovotestis, containing both ovarian and testicular tissue, or patients may have an ovary on one side and testis on the other. Infants may have a mixture of Mullerian and Wolffian structures internally, and ambiguous genitalia externally, although some patients have entirely female external genitalia. Diagnosis is guided by biopsy identifying ovarian and testicular tissues.15 Treatment of these individuals is complicated and involves removing dysgenetic testicular tissue, and ultimately proceeding with gender assignment.5 DISORDERS OF GLUCOSE HOMEOSTASIS Hypoglycemia of the newborn is a common problem, and its symptoms in neonates range from irritability to hypothermia to seizures. One of the clinical challenges is determining the circumstances under which a neonate may be hypoglycemic, when to check a blood glucose level, and what level is concerning. There is considerable debate regarding what glucose concentrations define hypoglycemia in the neonate; for practical purposes, we use ≤45 mg/dL.16 When infants present with recurrent hypoglycemia, blood work obtained while the infant is hypoglycemic is crucial to making the diagnosis (see Figure 2). There are many different causes of hypoglycemia, ranging from transient selfresolving processes to severe lifelong conditions. Transient hypoglycemia may be seen in children born to diabetic mothers or mothers using beta-blockers, and in children born small for gestational age (SGA). More persistent hypoglycemia may be caused by hypopituitarism and disorders of glycogenolysis, gluceoneogensis, lipolysis, and fatty acid oxidation.1 The most common cause of persistent neonatal hypoglycemia is congenital hyperinsulinism (estimated incidence of 1:50,000 live births), which can cause severe neurologic sequelae if not managed appropriately. The diagnosis is based on blood work obtained while the child is hypoglycemic, and includes the findings of low fatty acid levels, low PEDIATRIC ANNALS 42:5 | MAY 2013 CM E plasma ketone levels, and an increase in blood glucose levels in response to glucagon.17 Immediate treatment involves stabilizing glucose levels, frequently with high dextrose-containing intravenous fluids, while long-term management may include various medications like diazoxide, octreotide, or ultimately a partial or total pancreatic resection.18 Alternatively, hyperglycemia in the newborn period can be caused by neonatal diabetes (estimated incidence of 1 in 100,000 newborns).19 There are transient forms of neonatal diabetes, which typically resolve within 3 to 6 months of life, and permanent lifelong forms. The cellular defects noted in transient versus permanent neonatal diabetes appear to be different, but the clinical presentation of these two disorders is similar. Neonates present with intrauterine growth restriction, likely secondary to decreased levels of the growth factor insulin. They also have polyuria, failure to thrive, and can present with diabetic ketoacidosis.1 Treatment varies depending on the cause, but is typically with subcutaneous insulin. However, some forms of neonatal diabetes are amenable to oral sulfonylurea therapy.20 DISORDERS OF THE THYROID GLAND Thyroid dysfunction in the neonate can be broken down into hypothyroidism and hyperthyroidism. These categories can be further divided into transient versus permanent dysfunction. Transient hypothyroidism is defined by low thyroxine (T4) and elevated TSH concentrations that ultimately return to normal.21 Transient hypothyroidism can occur secondary to iodine deficiency, excess maternal anti-thyroid drugs reaching the fetus in utero, and transplacental passage of maternal thyrotropin receptor-blocking antibodies.22 In North America, transient hypothyroidism is rare, about 1 in 50,000, but is increased in countries with iodine deficiency. It PEDIATRIC ANNALS 42:5 | MAY 2013 Figure 2. Hypoglycemia evaluation in an infant. 3-OHB = 3-hydroxybutyrate; AA = acetoacetate; FFA = free fatty acid. *The physical examination or diagnostic imaging studies should demonstrate hepatomegaly. (Adapted from Fuhrman30) also appears to be increased in preterm infants as compared to term infants.21,23 Congenital hypothyroidism can be divided into two categories, primary (due to defects of the thyroid gland itself) or secondary (due to defects in the central nervous system), with primary being much more common. Thyroid dysgenesis (1:4,000) accounts for about 85% of primary congenital hypothyroidism, and is most commonly due to an ectopic thyroid gland, followed by thyroid aplasia and hypoplastic thyroid.24 The majority of cases of thyroid dysgenesis are sporadic.24 The remaining causes of primary congenital hypothyroidism are mostly secondary to thyroid dyshormonogenesis (1:40,000).1 Even rarer causes of primary hypothyroidism include errors in metabolism and transport of thyroid hormone, and defects in the thyroid hormone receptor. The most common cause of secondary hypothyroidism is hypopituitarism (1:100,000).1 Newborn screening has greatly aided in the prompt diagnosis of congenital hypothyroidism. If an infant is found to have an abnormal newborn screen, they should be immediately referred to a pediatric endocrinologist, as the goal is to treat as soon as possible due to the known importance of thyroid hormone on brain development.21 Importantly, the newborn screen in most states is designed to identify primary hypothyroidism; as such, cases of hypothyroidism due to hypopituitarism may not be identified. The American Academy of Pediatrics has created screening and treatment guidelines to facilitate the identification and treatment of these infants. Alternatively, hyperthyroidism in the neonatal period is rare, with the most common etiology being maternal Graves’ disease.25 Neonatal hyperthyroidism secondary to maternal Graves’ disease is due to the transplacental passage of maternal TSH receptor-stimulating antibodies (TSA) to the fetus. The most common symptom seen in the neonate is tachycardia, which can aid in diagnosis of this condition. The at-risk fetus can be monitored with maternal blood testing (TSA levels), fetal ultraHealio.com/Pediatrics | 71 CM E sound assessing for presence of goiter, and fetal heart rate monitoring if necessary.1,25 Treatment involves medical management of the symptoms and antithyroid drugs if needed. The disease course is self-limited (3 weeks to 12 weeks) and improves as the maternal antibodies are cleared.1 DISORDERS OF CALCIUM HOMEOSTASIS Calcium, phosphorous, and magnesium are carefully regulated in the human body. During pregnancy, the mother provides calcium and phosphorous to the fetus, which promotes skeletal development and tissue growth. After birth, the neonate is responsible for independent calcium homeostasis through a combination of calcium intake, the mobilization of calcium stores within the body, parathyroid hormone (PTH), vitamin D, and calcitonin. In the first few days of life, there is a drop in calcium concentrations followed by an acute compensatory increase in PTH, which then increases calcium levels.1,26 Calcium and phosphorus concentrations then gradually decline through 18 months of age. There are several systems in place to regulate calcium homeostasis, and errors in these mechanisms can cause both hypo- and hypercalcemia. Hypocalcemia in the neonate is often defined as a total calcium level < 7.5 mg/dL or an ionized calcium level < 1.20 mmol/L. Hypocalcemia may be asymptomatic, but may also present with multiple symptoms including, but not limited to, seizures, apnea, cyanosis, and arrhythmias. In the neonatal period, hypocalcemia is typically divided into early-onset (within the first 4 days of life) and late-onset (5-10 days after birth).27 Early-onset hypocalcemia may be attributed to prematurity and is also common in SGA infants. It is also seen in infants whose mothers have hyperparathyroidism or diabetes mellitus, as well as in infants with low birth weight, 72 | Healio.com/Pediatrics respiratory distress, and/or sepsis. From a mechanistic standpoint, early-onset hypocalcemia may be due to suppression of PTH, continued release of calcitonin, or hypomagnesemia.1 Alternatively, late-onset hypocalcemia can be caused by hypoparathyroidism (eg, DiGeorge syndrome and gain-of-function Hypocalcemia may present with multiple symptoms including, but not limited to, seizures, apnea, cyanosis, and arrhythmias. mutations in the calcium sensing receptor gene), pseudohypoparathyroidism, vitamin D deficiency, and the ingestion of high phosphorous formula.27 Evaluation includes a careful history and physical examination, initial laboratory studies determining total and ionized calcium, PTH level, vitamin D level, electrolytes, complete blood count, and urinary calcium, while treatment is aimed at symptom management and normalization of the serum calcium.1 Hypercalcemia in the neonate is defined as a total calcium level > 10.8 mg/dL; however significant symptoms are often not seen until a total calcium level of >12 mg/dL or an ionized calcium level >1.50 mmol/L is reached.28 Symptoms of hypercalcemia vary, and include anorexia, gastrointestinal reflux, nephrocalcinosis, lethargy, and seizures.1,28 There are numerous causes of hypercalcemia in the neonate, ranging from increased calcium absorption, to idiopathic hypercalcemia, to potentially life-threatening neonatal severe hyperparathyroidism.28 Infants born to mothers with hypocalcemia due to hypoparathyroidism or pseudohypoparathyroidism are also at risk. In general, hypercalcemia with normal PTH and low urine calcium levels is likely secondary to familial hypercalciuric hypercalcemia. Etiologies of elevated calcium and low PTH concentrations include increased calcium absorption, hypervitaminosis D, William’s syndrome, and subcutaneous fat necrosis. Alternatively, high PTH levels with hypercalemia indicates neonatal hyperparathyroidism.28 Evaluation of the etiology of hypercalcemia can be detailed, but should start with electrolytes (including calcium, magnesium, and phosphorus), calcitriol, vitamin D, and PTH levels. Treatment is aimed at normalizing calcium levels and managing symptoms.1 CONCLUSION The number of endocrine disorders that exist is vast, but many are uniquely relevant just to the newborn or to infants in the perinatal period. As such, the purpose of this review is to highlight some of the more common endocrinopathies that general pediatricians or neonatologists may encounter. Moreover, since the field of pediatric endocrinology continues to evolve and our understanding of the pathogenesis of certain endocrine disorders and their treatment continues to improve, consultation with a pediatric endocrinologist for any child with an endocrinopathy is always recommended. REFERENCES 1. Sperling M. Pediatric Endocrinology, 3rd ed. Philadelphia, PA: WB Saunders; 2008. 2. Mehta A, Dattani M. Developmental disorders of the hypothalamus and pituitary gland associated with congenital hypopituitarism. Best Pract Res Clin Endocrinol Metab. 2008;22(1):191-206. 3. Knepper MA. Molecular physiology of urinary concentrating mechanism: regulation of aquaporin water channels by vasopressin. Am J Physiol. 1997;272(1 Pt 2):F3-F12. 4. Wang LC, Cohen ME, Duffner PK. Etiologies of central diabetes insipidus in children. Pediatr Neurol. 1194;11(4):273-277. 5. Pescovitz O, Eugster E, eds. Pediatric Endocrinology: Mechanisms, Manifestations, and Management. Philadelphia, PA: Lippincott Williams & Wilkins; 2004. 6. Ranadive S, Rosenthal S. Pediatric endo- PEDIATRIC ANNALS 42:5 | MAY 2013 CM E crinology pediatric disorders of water balance. Endocrinol Metab Clin North Am. 2011;58(5):1271-1280. 7. Speiser P, White P. Congenital adrenal hyperplasia. N Engl J Med. 2003;349(8):776-788. 8. Shozu M, Akasofu K, Harada T, Kubota Y. A new cause of female pseudohermaphroditism: placental aromatase deficiency. J Clin Endocrinol Metab. 1191;72(3):560-566. 9. Kirk JM, Perry LA, Shand WS, Kirby RS, Besser GM, Savage MO. Female pseudohermaphroditism due to a maternal adrenocortical tumor. J Clin Endocrinol Metab. 1990;70(5):1280-1284. 10. Grumbach MM, Ducharme JR, Moloshok RE. On the fetal masculinizing action of certain oral progestins. J Clin Endocrinol Metab. 1959;19(11):1369-1380. 11. Piersma D, Verhoef-Post M, Berns E, Themmen A. LH receptor gene mutations and polymorphisms: an overview. Mol Cell Endocrinol. 2007;260-262:282-286. 12. Bose HS, Sugawara T, Strauss JF 3rd, Miller WL. The pathophysiology and genetics of congenial lipoid adrenal hyperplasia. N Engl J Med. 1996;335(25):1870-1878. 13. Wilson JD, Griffin JE, Russell DW. Steroid 5 alpha-reductase 2 deficiency. Endocr Rev. 1993;14(5):577-593. 14. Quigley CA, De Bellis A, Marschke KB, el- PEDIATRIC ANNALS 42:5 | MAY 2013 Awady MK, Wilson EM, French FS. Androgen receptor defects: historical, clinical, and molecular perspectives. Endocr Rev. 1995;16(3):271-321. 15. Kousta E, Papathanasiou A, Skordis N. Sex determination and disorders of sex development according to the revised nomenclature and classification in 46,XX individuals. Hormones (Athens). 2010;9(3):218-231. 16. Cornblath M, Hawdon JM, Williams AF, et al. Controversies regarding definition of neonatal hypoglycemia: suggested operational thresholds. Pediatrics. 2000;105(5):1141-1145. 17. Huopio H, Shyng SL, Otonkoski T, Nichols CG. K(ATP) channels and insulin disorders. Am J Physiol Endocrinol Metab. 2002;283(2):E207-E216. 18. Thornton PS, Alter CA, Katz LE, Baker L, Stanely CA. Short- and long-term use of octrotide in the treatment of congenital hyperinsulinism. J Pediatr. 1993;123(4):637-643. 19. Sperling MA. ATP-sensitive potassium channels — neonatal diabetes mellitus and beyond. N Engl J Med. 2006;355(5):507-510. 20. Pearson ER, Flechtner I, Njolstad PR, et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med. 2006;355(5):467-477. 21. AAP, Rose SR, Section on Endocrinology and Committee on Genetics. Update of newborn screening and therapy for congenital hypo- thyroidism. Pediatrics. 2006;117(60):22902303. 22. Parks JS, Lin M, Grosse SD, et al. The impact of transient hypothyroidism on the increasing rate of congenital hypothyroidism in the United States. Pediatrics. 2010;125(Suppl 2):S54-S63. 23. Delange F, Dalhem A, Bourdoux P, et al. Increased risk of primary hypothyroidism in perterm infants. J Pediatr. 1984;105(30:462-469. 24. LaFranchi SH. Approach to the diagnosis and treatment of neonatal hypothyroidism. J Clin Endocrinol Metab. 2011;96(10):2959-2967. 25. Zimmerman D. Fetal and neonatal hyperthyroidism. Thyroid. 1999;9(7):727-733. 26. Kovacs CS, Kronenberg HM. Maternal-fetal calcium and bone metabolism during pregnancy, puerperium, and lactation. Endocr Rev. 1997;18(6):832-872. 27.Thomas TC, Smith JM, White PC, Adhikari S. Transient neonatal hypocalcemia: presentation and outcomes. Pediatrics. 2012;129(6):e1461-e1467. 28. Soriano JR. Neonatal hypercalcemia. J Nephrol. 2003;16(4):606-608. 29. Oberfield SE, Sopher AB, Gerken AT. Approach to the girl with early onset pubic hair. J Clin Endocrinol Metab. 2011;96(6):1610-1622. 30. Fuhrman BP. Pediatric Critical Care. 4th Ed. Philadelphia, PA: WB Saunders; 2011. Healio.com/Pediatrics | 73