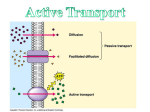
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Molecular neuroscience wikipedia , lookup
Protein adsorption wikipedia , lookup
Protein–protein interaction wikipedia , lookup
Chemical synapse wikipedia , lookup
Two-hybrid screening wikipedia , lookup
Signal transduction wikipedia , lookup
Western blot wikipedia , lookup
Cell membrane wikipedia , lookup
SNARE (protein) wikipedia , lookup
Cell-penetrating peptide wikipedia , lookup
Endomembrane system wikipedia , lookup
Independent Meeting held at the University of Edinburgh, March 23–25, 2003. Edited by D. Apps (Edinburgh). Supported by The Wellcome Trust, The International Society for Neurochemistry, The Physiological Society, The Biochemical Society and The International Union of Biochemistry and Molecular Biology. Visualizing membrane trafficking using total internal reflection fluorescence microscopy V. Beaumont1 MRC Laboratory of Molecular Biology, Hills Road, Cambridge CB2 2QH, U.K. Abstract There is a dizzying array of fluorescent probes now commercially available to monitor cellular processes, and advances in molecular biology have highlighted the ease with which proteins can now be labelled with fluorophores without loss of functionality. This has led to an explosion in the popularity of fluorescence microscopy techniques. One such specialized technique, total internal reflection fluorescence microscopy (TIR-FM), is ideally suited to gaining insight into events occurring at, or close to, the plasma membrane of live cells with excellent optical resolution. In the last few years, the application of TIR-FM to membrane trafficking events in both non-excitable and excitable cells has been an area of notable expansion and fruition. This review gives a brief overview of that literature, with emphasis on the study of the regulation of exocytosis and endocytosis in excitable cells using TIR-FM. Finally, recent applications of TIR-FM to the study of cellular processes at the molecular level are discussed briefly, providing promise that the future of TIR-FM in cell biology will only get brighter. Introduction to total internal reflection fluorescence microscopy (TIR-FM) TIR-FM is a technique that specifically illuminates fluorophores in a thin optical section above the interface between two media with different diffractive indices. To achieve this, laser light is directed obliquely at the interface between two media from a high (n1 ) to a low (n2 ) diffractive index with an incident angle greater than the critical angle (θ c ) of total internal reflection (TIR). θ c is given by: θc = sin−1 \(n2 /n1 ) (1) Under these conditions, laser light is totally internally reflected at the interface. Even so, some of the light still penetrates the medium of lower diffractive index as an electromagnetic field called the ‘evanescent wave’. A key characteristic of the evanescent wave is that it propagates parallel Key words: endocytosis, evanescent wave microscopy, exocytosis. Abbreviations used: EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; FRET, fluorescence resonance energy transfer; (e)GFP, (enhanced) green fluorescent protein; PKC, protein kinase C; SNARE, soluble N-ethylmaleimide-sensitive factor attachment protein receptor; TIR, total internal reflection; TIR-FM, total internal reflection fluorescence microscopy. 1 e-mail [email protected] to the interface, vanishing exponentially with distance. The decay length (d) of the evanescent wave along the depth of field depends on the incident angle (θ), the wavelength of the excitation beam (λ) and the diffractive indices of both media: d = λ/[4π (n1 2 · sin2 \θ − n2 2 )] (2) The decay of the evanescent field with depth from the interface z is described as: I(z) = I0 e−z/d (3) Evanescent field penetration depths (the distance from the interface whereby intensity decreases to I0 /e) of <100 nm can be readily achieved from TIR at glass/aqueous interfaces, providing an opportunity to selectively excite fluorophores in an optical slice with the dimensions of thin electron microscopy. This offers a 5–10-fold advantage over the optical resolution achievable with confocal microscopy (≈500 nm). If cells can be grown on, or temporarily adhered to, glass coverslips, imaging of fluorophores at, or immediately below, the plasma membrane can be achieved in live cells. In pioneering work by Daniel Axelrod, this technique was C 2003 Biochemical Society Molecular Mechanisms of Exocytosis and Endocytosis Independent Meeting Molecular Mechanisms of Exocytosis and Endocytosis 819 820 Biochemical Society Transactions (2003) Volume 31, part 4 first applied to the study of live cells, primarily to investigate cell–substrate adhesions [1]. There are numerous excellent reviews describing the TIR-FM technique in detail, ranging from detailed explanations of the physics of evanescent field generation [2–7] to practical implementation of TIR-FM, and construction of either ‘through-the-prism’ or ‘through-theobjective’ TIR-F microscopes [8–10]. In ‘prism-type’ microscopes, incident light from a laser strikes a prism that is coupled to the coverslip by glycerol or another liquid of matching refractive index, and the light emitted by fluorophores excited within the evanescent field is collected by a high-numerical-aperture water immersion lens. This technique allows fine control of the angle of incidence, which is useful if one wishes to change the penetration depth of the evanescent field. This can be achieved with high precision by using acousto-optical detectors to rapidly alter the incident angle of the laser light, and has been used to track fluorophore motion into deeper regions of cells [11–13]. In through-the-objective TIR-FM, the incident laser light is directed at an appropriate angle directly through the objective of an inverted microscope. This imposes restraints on the angle achievable from the incident laser light, and hence on the resultant evanescent field depth. In practice, incident angles exceeding θ c for a glass/water interface are only achieved with objectives with numerical apertures of >1.4, and d still cannot be controlled as accurately as in prism-type TIR-FM. However, this small loss in precision allows greater accessibility to the specimen – through-theobjective set-ups leave the top surface unobstructed, allowing TIR-FM to be easily combined with other techniques. TIRFM has been used in combination with patch-clamping, providing precise electrical control of individual cells, and a synchrony between electrical and optical output [14–17]. Laser-trap forcemetry has also been combined with TIRFM in bovine chromaffin cells to investigate the directional flow of an exocytic burst, as well as the differential force of expulsion of secretory products [18]. Exocytosis and granule dynamics in endocrine cells Observing the motion and fusion of secretory granules docked beneath the plasma membrane of isolated chromaffin cells heralded the beginnings of using TIR-FM to explore the molecular steps controlling exocytosis. By taking advantage of the fact that certain fluorescent acidotropic dyes (such as Acridine Orange) accumulate in acidic compartments of cells, such as granules, it was possible to identify individual fluorescing secretory granules within the evanescent field. Detailed information about granule docking and mobility was obtained by several groups [12,19–21], which showed that granules above the plasma membrane are free to move, albeit in a restricted manner, and can approach the membrane in directed trajectories, where they subsequently become relatively immobile (or docked). While the docking step was reversible (granules could move away from the plasma mem C 2003 Biochemical Society brane after remaining resident for 10s of seconds), docked granules were shown to fuse with the plasma membrane in response to stimulation by a depolarizing solution, releasing their contents along with a puff of fluorescent dye. These studies opened up the possibility that the regulation of pre-fusion steps could be studied in live cells in a noninvasive manner. In particular, the effects of cytoskeletal modification using actin depolymerizers [22] and the effects of neuromodulators on the regulation of granule mobility were assessed directly. For instance, in bovine chromaffin cells, Tsuboi et al. [23] showed that long-term (>30 min) elevation of protein kinase C (PKC) activity approximately doubled the morphologically docked pool of granules, consistent with the earlier idea that PKC results in an increase in the supply of granules to the plasma membrane, which may explain its actions as a potent sectretagogue, despite its concomitant inhibition of calcium influx through voltagegated calcium channels [24]. Interestingly, prior actin depolymerization of chromaffin cells rendered PKC activation unable to increase the docked granule pool, suggesting that PKC may influence granule docking by altering the cortical actin network in these cells [25–27]. However, the fusion machinery itself has also been proposed as a direct target in the PKC-dependent enhancement of exocytosis [28–30]. In MIN6 and pancreatic β-cells, granules containing insulin are released in response to elevated external glucose concentrations. Using TIR-FM, recruitment of PKC to the plasma membrane in response to glucose was shown by constructing fusion constructs of enhanced green fluorescent protein (eGFP) with PKC isoforms [31], suggesting that PKC becomes well placed to regulate granule fusion, perhaps by a direct effect on the fusion machinery. Other investigators have shown a preferential release of the docked granule pool containing insulin–GFP in MIN6 β-cells [32], and have furthermore shown that this exocytosis could be inhibited by introduction of the syntaxin H3 domain [to interfere with SNARE complex formation, where SNARE is defined as soluble N-ethylmaleimide-sensitive factor attachment protein receptor], confirming that granule fusion (but not pre-docking) is a SNARE-dependent process [33]. TIR-FM in PC-12 cells has also been used to study organelle dynamics and the regulation of exocytosis. In cells overexpressing a hybrid neuronal calcium sensor-1 protein (a homologue of the non-mammalian protein frequenin) fused to enhanced yellow fluorescent protein, synaptic-like microvescicles, but not granules, were shown to undergo an enhanced rate of both regulated and constitutive exocytosis [34]. Investigation of the coupling between exocytosis and endocytosis has been assisted by the use of dual-colour TIR-FM, whereby the proteins of interest, labelled with different variants of GFPs, can be distinguished spectrally. The emitted light from the two fluorophores is split by the use of appropriate dichroics and filters, and each image is illuminated at different areas on the chip of a CCD camera [as achieved with the W-view optical system (A4314; Hamamatsu Photonics), the MultiSpec MicroImager (Optical Insights) or the Cairn Optosplit]. This allows Molecular Mechanisms of Exocytosis and Endocytosis the simultaneous imaging of the spatio–temporal dynamics of protein interactions. In PC-12 cells, synaptobrevin (tagged with DsRed) and dynamin (tagged with eGFP) have been imaged simultaneously. By capturing the exact site and moment of exocytosis in response to electrical stimulation (by watching the rapid and transient increase in synaptobrevin–eGFP fluorescence), and comparing this to the foci of endocytic activity, the authors showed that clusters of DsRed–dynamin appeared randomly at the plasma membrane following stimulation, gliding laterally prior to disappearance (see also [35]). It was shown that more than 70% of the exocytic responses of synaptobrevin had no immediate and specific dynamin counterpart at the same site. Thus the stimulus-dependent recruitment of dynamin, and subsequent ‘x–y’ movement, was interpreted as a ‘scanning’ of the membrane for invaginated pits. This supports the notion that endocytosis is not well coupled, at least temporally, to regulated exocytosis in neuroendocrine cells [36]. On the same theme, another recent study, again in PC12 cells, utilized dual-colour TIR-FM to explore the fate of post-exocytosed granules, and inferred a direct spatial coupling of exocytosis with granule recapture (presumably representing the endocytic event) [37]. Using the relatively soluble granule peptide neuropeptide Y tagged with cyan fluorescent protein (to detect the fusion of granules and release of neuropeptide Y), in conjunction with either the larger soluble granule protein tissue plasminogen activator tagged with the yellow fluorescent protein Venus, or the granule membrane protein phogrin tagged with DsRed, the authors investigated the relationship between fusion and post-exocytic membrane handling. They showed that, following exocytosis, the membrane protein phogrin almost entirely failed to disperse (surprisingly unlike the rapid dispersal of synaptobrevin [36]), and that granules kept their shape for several minutes following exocytosis, resealing and re-acidifying at the same locus as the exocytic event itself. The sequence of protein recruitment to the plasma membrane controlling the endocytic phase of clathrindependent constitutive membrane retrieval has also been scrutinized in fibroblasts [38]. Cells stably expressing GFPlabelled β-actin or dynamin-1–eGFP were transfected with a clathrin-light-chain–DsRed fusion protein. The data showed that, after remaining stationary for 10s of seconds at the plasma membrane, clathrin-coated pits were internalized immediately after a brief burst of dynamin recruitment, and that movement of the clathin-coated structure away from the membrane was accompanied by a transient assembly of actin. Thus the authors suggest that dynamin may provide the trigger, and actin the force, for movement of clathrin-coated structures into the cytosol. Exocytosis and vesicle dynamics in neurons Real-time optical monitoring of synaptic vesicle recycling in neurons became possible with the advent of the fluorescent styryl dyes such as FM1-43 (for a review, see [39]). These dyes partition into lipid membranes and become trapped in recycled vesicles on endocytosis. Further stimulation of synaptic transmission causes preloaded vesicles to release their dye during subsequent rounds of exocytosis. Using epifluorescence microscopy, this is observed as a loss of dye from synaptic terminals. The relative sparsity of TIR-FM studies in neurons compared with neuroendocrine cells is related in part to the difficulty in finding a suitable preparation to study – namely synaptic terminals that are large, and which adhere closely to glass when either acutely dissociated or cultured. These criteria have been well met by the ON-type bipolar neurons from the goldfish retina, whose giant synaptic terminals, ≈10 µm in diameter, are packed with synaptic vesicles, and adhere well to glass in the absence of an associated postsynaptic cell following acute enzymic dissociation. With the excellent background fluorescence rejection achievable with TIR-FM, it became possible for the first time to image single synaptic vesicle pre-fusion and fusion events at these presynaptic sites [15]. ‘Docked’ vesicles (stationary for >0.5 s) were apparent as punctate fluorescence superimposed on a background of FM1-43 staining of the plasma membrane. The authors showed that there were also preferred sites at which vesicles attached to the plasma membrane, and furthermore that vesicles that were attached fused preferentially and rapidly on inititation of a depolarizing stimulus (seen as a brightening and lateral spread of FM1-43), while vesicles recruited to the plasma membrane contributed to further exocytosis at later time points. In agreement with studies on neuroendocrine cells, Zenisek et al. [15] were able to determine that the docking, or ‘capture’, of vesicles at the plasma membrane was indeed reversible. The time required for docked vesicles to reach exocytic competence (the socalled priming step) was estimated as ≈210 ms. In a later paper, the same authors also provided detailed information about the nature of the exocytic event itself, providing evidence that, on depolarization, FM1-43-stained synaptic vesicles fuse completely enough with the plasma membrane to allow the diffusion of dye into the surrounding membrane in entirety. This finding was interpreted as evidence of fusion occurring by the classical Heuser and Reese view of exo-endocytosis – namely a flattening of the vesicle with the plasma membrane on fusion, prior to endocytosis [40], rather than by the opening of a transient fusion pore (the ‘kiss-and-run’ exocytosis) first suggested by Meldolesi and Ceccarelli [41]. Further work on goldfish bipolar neurons will no doubt continue to provide exciting advances in our understanding of the regulation and modulation of synaptic vesicle recycling. However, long-term culture of these neurons, and transfection with fluorescently tagged proteins, has proved problematic, and may ultimately limit the usefulness of this preparation in application of the molecular biology tools so complimentary to the TIR-FM studies performed in other cells. However, work in progress (V. Beaumont, unpublished work) suggests that mouse bipolar neurons are equally well suited to TIR-FM studies, and no doubt other preparations will be discovered in time. Investigation of pre-fusion events C 2003 Biochemical Society 821 822 Biochemical Society Transactions (2003) Volume 31, part 4 and synaptic vesicle cycling, with the potential for singlemolecule resolution, in living mammalian neurons would represent a unique functional assay for many of the synaptic proteins already identified, and which can be altered using transgenic technology. From identifying single vesicles to single molecular events What does the future hold for cell biology using TIR-FM? An exciting advance is the achievement of single-molecule optical detection. The largest problem to overcome in singlemolecule fluorescence studies is typically the necessity to reduce background noise in order to amplify the signal-tonoise ratio of single fluorophores. The most effective way to reduce this background is to limit the excitation volume to the region of interest. In 1995, Funatsu et al. [42] demonstrated that the small excitation volume achievable with TIR-FM was ideal for this purpose. Fluorophores for single-molecule detection must be relatively robust, having a large molar absorption coefficient and a high quantum yield, and being relatively resistant to photobleaching. Proteins conjugated to Alexa488, tetramethylrhodamine, Cy3, Alexa598 and Cy5 have successfully met these criteria and have been used to visualize singlemolecule reactions in living cells. Binding of epidermal growth factor (EGF) labelled in a 1:1 ratio with Cy3 to EGF receptors (EGFRs) and subsequent dimerization of the EGF–EGFR complexes has been demonstrated at the single-molecule level in live HeLa cells [43]. The kinetics of dissociation between a chemoattractant and its membrane receptor have also been studied using TIR-FM. Dictoyostelium amoebae perform chemotaxis in response to cAMP. To elucidate how cells sense gradients of cAMP, Ueda et al. [44] synthesized Cy3-labelled cAMP and observed its binding to individual receptors in cells undergoing chemotactic migration. In addition, proteins tagged with GFP and yellow fluorescent protein have also been imaged at the single-molecule level. For instance, single GFP-labelled kinesin molecules have been imaged travelling along Cy3-labelled microtubules to determine how various microtubule-associated proteins interfere with the attachment of these molecular motors [45]. The potential to detect the intermolecular interactions of biomolecules at the single-molecule level using fluorescence resonance energy transfer (FRET) combined with TIR-FM has been elegantly demonstrated by Suzuki and co-workers [46]. They devised a method whereby the fluorescence spectrum of a single molecule can be assayed using TIR-FM by passing the emitted light through a dispersion prism for spectroscopy. Single-pair FRET was demonstrated between Rhodamine Red (labelling the essential light chain of myosin) and Cy5 (labelling the myosin heavy chain) in a reconstructed double-labelled myosin motor domain, immobilized on a quartz coverslip. The structural rearrangement of a single protein has also been detected. Tetramethylrhodamine was attached to C 2003 Biochemical Society specific cysteine residues in the voltage-sensing S4 segment of Shaker K+ channels expressed in the plasma membrane of oocytes [14]. Fluorescence intensity changed in response to the voltage-sensitive movements of the protein channel around the dye, as depolarization drove the S4 segment from the resting to the activated conformation. Conclusions The compatibility of TIR-FM with other techniques, in particular voltage clamping, means that it should be possible to relate directly the structural rearrangements of individual proteins, or the spatio–temporal co-ordination of protein interactions, to physiological cell signalling events measured simultaneously. Advances in molecular biology and protein structure are rapidly uncovering the molecular players of exocytosis and endocytosis. Clearly, one of the key objectives in the future is to analyse comprehensively how these molecular machines operate together to mediate the complex and highly regulated process of synaptic vesicle fusion and recycling, and TIR-FM techniques seem poised to do exactly that. Furthermore, there are clear advantages of being able to study these interactions ‘in situ’, where one does not have to worry about the near-impossible task of reconstituting appropriate cellular conditions. V.B. is supported financially by a postdoctoral fellowship from the Medical Research Council, U.K. References 1 Axelrod, D. (1981) J. Cell Biol. 89, 141–145 2 Axelrod, D., Thompson, N.L. and Burghardt, T.P. (1983) J. Microsc. 129, 19–28 3 Axelrod, D., Burghardt, T.P. and Thompson, N.L. (1984) Annu. Rev. Biophys. Bioeng. 13, 247–268 4 Axelrod, D. (1989) Methods Cell Biol. 30, 245–270 5 Steyer, J.A. and Almers, W. (2001) Nat. Rev. Mol. Cell Biol. 2, 268–275 6 Thompson, N.L. and Lagerholm, B.C. (1997) Curr. Opin. Biotechnol. 8, 58–64 7 Toomre, D. and Manstein, D.J. (2001) Trends Cell Biol. 11, 298–303 8 Oheim, M. (2001) Lasers Med. Sci. 16, 149–158 9 Oheim, M. (2001) Lasers Med. Sci. 16, 159–170 10 Axelrod, D. (2001) Traffic 2, 764–774 11 Rohrbach, A. (2000) Biophys. J. 78, 2641–2654 12 Oheim, M., Loerke, D., Stuhmer, W. and Chow, R.H. (1999) Eur. Biophys. J. 28, 91–101 13 Mathur, A.B., Truskey, G.A. and Reichert, W.M. (2000) Biophys. J. 78, 1725–1735 14 Sonnleitner, A., Mannuzzu, L.M., Terakawa, S. and Isacoff, E.Y. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 12759–12764 15 Zenisek, D., Steyer, J.A. and Almers, W. (2000) Nature (London) 406, 849–854 16 Zenisek, D., Steyer, J.A., Feldman, M.E. and Almers, W. (2002) Neuron 35, 1085–1097 17 Ide, T., Takeuchi, Y., Aoki, T. and Yanagida, T. (2002) Jpn. J. Physiol. 52, 429–434 18 Tsuboi, T., Kikuta, T., Sakurai, T. and Terakawa, S. (2002) Biophys. J. 83, 172–183 19 Steyer, J.A., Horstmann, H. and Almers, W. (1997) Nature (London) 388, 474–478 20 Oheim, M., Loerke, D., Stuhmer, W. and Chow, R.H. (1998) Eur. Biophys. J. 27, 83–98 21 Steyer, J.A. and Almers, W. (1999) Biophys. J. 76, 2262–2271 22 Oheim, M. and Stuhmer, W. (2000) Eur. Biophys. J. 29, 67–89 Molecular Mechanisms of Exocytosis and Endocytosis 23 Tsuboi, T., Kikuta, T., Warashina, A. and Terakawa, S. (2001) Biochem. Biophys. Res. Commun. 282, 621–628 24 Gillis, K.D., Mossner, R. and Neher, E. (1996) Neuron 16, 1209–1220 25 Vitale, M.L., Rodriguez Del Castillo, A. and Trifaro, J.M. (1992) Neuroscience 51, 463–474 26 Trifaro, J., Rose, S.D., Lejen, T. and Elzagallaai, A. (2000) Biochimie 82, 339–352 27 Sasakawa, N., Ohara-Imaizumi, M., Okubo, S., Hosaka, S., Hayashi, M. and Kumakura, K. (2002) Ann. N.Y. Acad. Sci. 971, 273–274 28 Barclay, J.W., Craig, T.J., Fisher, R.J., Ciufo, L.F., Evans, G.J., Morgan, A. and Burgoyne, R.D. (2003) J. Biol. Chem. 278, 10538–10545 29 Caohuy, H. and Pollard, H.B. (2002) J. Biol. Chem. 277, 25217–25225 30 Nagy, G., Matti, U., Nehring, R.B., Binz, T., Rettig, J., Neher, E. and Sorensen, J.B. (2002) J. Neurosci. 22, 9278–9286 31 Pinton, P., Tsuboi, T., Ainscow, E.K., Pozzan, T., Rizzuto, R. and Rutter, G.A. (2002) J. Biol. Chem. 277, 37702–37710 32 Ohara-Imaizumi, M., Nakamichi, Y., Tanaka, T., Ishida, H. and Nagamatsu, S. (2002) J. Biol. Chem. 277, 3805–3808 33 Ohara-Imaizumi, M., Nakamichi, Y., Nishiwaki, C. and Nagamatsu, S. (2002) J. Biol. Chem. 277, 50805–50811 34 Scalettar, B.A., Rosa, P., Taverna, E., Francolini, M., Tsuboi, T., Terakawa, S., Koizumi, S., Roder, J. and Jeromin, A. (2002) J. Cell Sci. 115, 2399–2412 35 Tsuboi, T., Zhao, C., Terakawa, S. and Rutter, G.A. (2000) Curr. Biol. 10, 1307–1310 36 Tsuboi, T., Terakawa, S., Scalettar, B.A., Fantus, C., Roder, J. and Jeromin, A. (2002) J. Biol. Chem. 277, 15957–15961 37 Taraska, J.W., Perrais, D., Ohara-Imaizumi, M., Nagamatsu, S. and Almers, W. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 2070–2075 38 Merrifield, C.J., Feldman, M.E., Wan, L. and Almers, W. (2002) Nat. Cell Biol. 4, 691–698 39 Angleson, J.K. and Betz, W.J. (1997) Trends Neurosci. 20, 281–287 40 Heuser, J.E. and Reese, T.S. (1973) J. Cell Biol. 57, 315–344 41 Meldolesi, J. and Ceccarelli, B. (1981) Philos. Trans. R. Soc. London B Biol. Sci. 296, 55–65 42 Funatsu, T., Harada, Y., Tokunaga, M., Saito, K. and Yanagida, T. (1995) Nature (London) 374, 555–559 43 Sako, Y., Minoghchi, S. and Yanagida, T. (2000) Nat. Cell Biol. 2, 168–172 44 Ueda, M., Sako, Y., Tanaka, T., Devreotes, P. and Yanagida, T. (2001) Science 294, 864–867 45 Seitz, A., Kojima, H., Oiwa, K., Mandelkow, E.M., Song, Y.H. and Mandelkow, E. (2002) EMBO J. 21, 4896–4905 46 Suzuki, Y., Tani, T., Sutoh, K. and Kamimura, S. (2002) FEBS Lett. 512, 235–239 Received 24 February 2003 C 2003 Biochemical Society 823