Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Oligonucleotide synthesis wikipedia , lookup
NADH:ubiquinone oxidoreductase (H+-translocating) wikipedia , lookup
Fatty acid synthesis wikipedia , lookup
Mitochondrion wikipedia , lookup
Basal metabolic rate wikipedia , lookup
Evolution of metal ions in biological systems wikipedia , lookup
Photosynthesis wikipedia , lookup
Nicotinamide adenine dinucleotide wikipedia , lookup
Nitrogen cycle wikipedia , lookup
Fatty acid metabolism wikipedia , lookup
Microbial metabolism wikipedia , lookup
Glyceroneogenesis wikipedia , lookup
Biosynthesis wikipedia , lookup
Adenosine triphosphate wikipedia , lookup
Oxidative phosphorylation wikipedia , lookup
Amino acid synthesis wikipedia , lookup
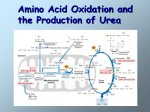
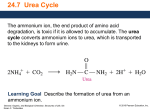
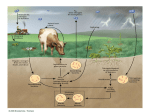
Urea cycle From Wikipedia, the free encyclopedia The urea cycle (also known as the ornithine cycle) is a cycle of biochemical reactions occurring in many animals that produces urea ((NH2)2CO) from ammonia(NH3). This cycle was the first metabolic cycle discovered (Hans Krebs and Kurt Henseleit, 1932), five years before the discovery of the TCA cycle. In mammals, the urea cycle takes place primarily in the liver, and to a lesser extent in the kidney. Function Organisms that cannot easily and quickly remove ammonia usually have to convert it to some other substance, like urea or uric acid, which are much less toxic. Insufficiency of the urea cycle occurs in some genetic disorders (inborn errors of metabolism), and in liver failure. The result of liver failure is accumulation of nitrogenous waste, mainly ammonia, which leads to hepatic encephalopathy. Reactions The urea cycle consists of five reactions: two mitochondrial and three cytosolic. The cycle converts two amino groups, one from NH4+ and one from Asp, and a carbon atom from HCO3−, to the relatively nontoxic excretion product urea at the cost of four "high-energy" phosphate bonds (3 ATP hydrolyzed to 2 ADP and one AMP). Ornithine is the carrier of these carbon and nitrogen atoms. Reactions of the urea cycle Step Reactants Products Catalyzed by Location 1 NH4+ + HCO3− + 2ATP 2 carbamoyl phosphate + ornithine citrulline + Pi OTC mitochondria 3 citrulline + aspartate + ATP argininosuccinate + AMP + PPi ASS cytosol 4 argininosuccinate Arg + fumarate ASL cytosol 5 Arg + H2O ornithine + urea ARG1 cytosol carbamoyl phosphate + 2ADP + Pi CPS1 mitochondria The reactions of the urea cycle 1 L-ornithine 2 carbamoyl phosphate 3 L-citrulline 4 argininosuccinate 5 fumarate 6 L-arginine 7 urea L-Asp L-aspartate CPS-1 carbamoyl phosphate synthetase I OTC Ornithine transcarbamoylase ASS argininosuccinate synthetase ASL argininosuccinate lyase ARG1 arginase 1 In the first reaction, NH4+ + HCO3− is equivalent to NH3 + CO2 + H2O. Thus, the overall equation of the urea cycle is: NH3 + CO2 + aspartate + 3 ATP + 2 H2O → urea + fumarate + 2 ADP + 2 Pi + AMP + PPi Since fumarate is obtained by removing NH3 from aspartate (by means of reactions 3 and 4), and PPi + H2O → 2 Pi, the equation can be simplified as follows: 2 NH3 + CO2 + 3 ATP + H2O → urea + 2 ADP + 4 Pi + AMP This process requires energy, but it is necessary to convert the toxic ammonia into non-toxic urea to transport it to the kidneys to remove the nitrogen waste. Note that reactions related to the urea cycle also cause the production of 2 NADH, so the urea cycle releases slightly more energy than it consumes. These NADH are produced in two ways: One NADH molecule is reduced by the enzyme glutamate dehydrogenase in the conversion of glutamate to ammonium and α-ketoglutarate. Glutamate is the non-toxic carrier of amine groups. This provides the ammonium ion used in the initial synthesis of carbamoyl phosphate. The fumarate released in the cytosol is converted to malate by cytosolic fumarase. This malate is then converted to oxaloacetate by cytosolic malate dehydrogenase, generating a reduced NADH in the cytosol. Oxaloacetate is one of the keto acids preferred by transaminases, and so will be recycled toaspartate, maintaining the flow of nitrogen into the urea cycle. The two NADH produced can provide energy for the formation of 4 ATP(cytosolic NADH provides only 1.5 ATP due to the glycerol-3-phosphate shuttle who transfers the electrons from cytosolic NADH to FADH2 and that gives 1.5 ATP), a net production of one high-energy phosphate bond for the urea cycle. However, ifgluconeogenesis is underway in the cytosol, the latter reducing equivalent is used to drive the reversal of the GAPDH step instead of generating ATP. The fate of oxaloacetate is either to produce aspartate via transamination or to be converted to phosphoenol pyruvate, which is a substrate to glucose. Regulation N-Acetylglutamic acid The synthesis of carbamoyl phosphate and the urea cycle are dependent on the presence of NAcGlu, which allosterically activates CPS1. NAcGlu is an obligate activator of Carbamoyl phosphate synthase[1]. Synthesis of NAcGlu by NAGS, is stimulated by both Arg, allosteric stimulator of NAGS, and Glu, a product in the transamination reactions and one of NAGS's substrates, both of which are elevated when free amino acids are elevated. So, Glu is not only a substrate for NAGS but also serves as an activator for the urea cycle. Substrate concentrations The remaining enzymes of the cycle are controlled by the concentrations of their substrates. Thus, inherited deficiencies in the cycle enzymes other than ARG1do not result in significant decrease in urea production (the total lack of any cycle enzyme results in death shortly after birth). Rather, the deficient enzyme's substrate builds up, increasing the rate of the deficient reaction to normal. The anomalous substrate buildup is not without cost, however. The substrate concentrations become elevated all the way back up the cycle to NH4+, resulting in hyperammonemia (elevated [NH4+]P). Although the root cause of NH4+ toxicity is not completely understood, a high [NH4+] puts an enormous strain on the NH4+-clearing system, especially in the brain (symptoms of urea cycle enzyme deficiencies include mental retardation and lethargy). This clearing system involves GLUD1 and GLUL, which decrease the2-oxoglutarate (2OG) and Glu pools. The brain is most sensitive to the depletion of these pools. Depletion of 2OG decreases the rate of TCAC, whereas Glu is both a neurotransmitter and a precursor to GABA, another neurotransmitter. [1](p.734) Pathology Anomalies of the urea cycle cause urea cycle disorders: ornithine transcarbamoylase deficiency Carbamoyl phosphate synthetase deficiency Argininosuccinic aciduria Argininemia Hyperornithinemia, hyperammonemia, homocitrullinuria syndrome (HHH syndrome, ornithine translocase deficiency) Lysinuric protein intolerance Citrullinemia N-Acetylglutamate synthase deficiency Most of them are associated with hyperammonemia. Additional images Urea cycle. Urea cycle colored.