Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Discovery and development of direct Xa inhibitors wikipedia , lookup
Discovery and development of ACE inhibitors wikipedia , lookup
Discovery and development of proton pump inhibitors wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Discovery and development of TRPV1 antagonists wikipedia , lookup
Drug discovery wikipedia , lookup
Drug interaction wikipedia , lookup
Drug design wikipedia , lookup
NMDA receptor wikipedia , lookup
5-HT2C receptor agonist wikipedia , lookup
Discovery and development of beta-blockers wikipedia , lookup
Psychopharmacology wikipedia , lookup
Toxicodynamics wikipedia , lookup
Nicotinic agonist wikipedia , lookup
Discovery and development of angiotensin receptor blockers wikipedia , lookup
Discovery and development of antiandrogens wikipedia , lookup
Neuropharmacology wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
CCR5 receptor antagonist wikipedia , lookup
5-HT3 antagonist wikipedia , lookup
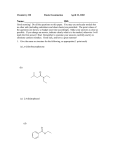
Chapter 11 Case Study Answer Conduct a thorough and mechanistic SAR analysis of the 3 therapeutic options in the case. 5-HT3 antagonists share a common structural motif: an aromatic ring, a hydrogen-accepting carbonyl moiety coplanar with the aromatic ring, and a relatively unhindered basic nitrogen atom all separated by a defined distance. The 1-azabicyclo[2.2.2]octane (quinuclidine) ring system of compound 1 is reminiscent of the tropane ring system (8-azabicyclo[3.2.1]octane) of cocaine, the drug that served as the template for the development of selective 5-HT3 antagonists. These two ring systems are known to provide high potency 5-HT3 antagonism. The restrained bicyclic ring ensures a sterically unrestricted amino nitrogen which is presumed to bind to the 5-HT3 receptor (a ligand-gated ion channel) in protonated form. The nature of the aromatic ring can vary, and substitution, when present, is generally limited to lipophilic and/or small groups. 5-HT3 antagonists have found a therapeutic niche in the prevention of cancer chemotherapy and/or radiation-induced nausea and vomiting (CINV). In addition to being a highly selective 5HT3 antagonist, compound 1 also binds persistently to its receptor. Some authors have estimated its affinity for 5-HT3 receptors as over 30 times that of “1st generation” antagonists without a concomitant increase in side effects (Aapro, Ther Clin Risk Manag 2007;3(6):1009-1020). With an elimination half-life of up to almost 2 days, compound 1 is retained in vivo for an extended period. It produces a prolonged therapeutic effect that is useful in treating both the acute and the delayed phases of CINV. It is slowly metabolized to two clinically inactive metabolites, the N-oxide and the 6-hydroxy analog. The hydroxylation reaction is stereospecific. CYP2D6 catalyzes the bulk of the biotransformation, but 2D6 phenotype does not impact the activity or side effect profile of this drug and the risk of drug-drug interaction is low. While some clinically significant cardiac toxicity has been observed with 5-HT3 antagonists, compound 1 is not among them. The increase in QTc interval produced by this agent has been deemed clinically insignificant and it is considered safe to use in patients with underlying cardiovascular disease. Compound 2 is another 5-HT3 antagonist that has replaced the azabicyclic ring system found in compound 1 with imidazole. These two ring systems are the most common and potent carriers for the required basic amino nitrogen atom. The aromatic ring is now indole and the Haccepting group is a ketone. These two groups are coplanar, as required, and the single sp2 hybridized atom separation of the carbonyl and benzo component of the indole ring is optimal. The indole and imidazole rings of compound 2 are substituted with small, lipophilic methyl groups, which honors steric restriction rules for these areas of the antagonist. While these differences in structure do not alter selectivity for the 5-HT3 receptor, compound 2 does not bind with the tenacity of compound 1. Compound 2 is vulnerable to inactivating metabolism, with CYP3A4 catalyzing the predominant indole hydroxylation reaction. Indole hydroxylation also occurs at C7 and both monophenols and the catechol are found in the serum of patients on this antiemetic. The 4 hour elimination half-life of this agent is the shortest of all of the drugs in its class. The lower binding affinity and more facile metabolic inactivation of compound 2 result in a much shorter duration of therapeutic action in the prevention of CINV compared to 1. Compound 2 is also known to prolong the QTc interval. In addition, while not anticipated based on mechanism or metabolic profile, compound 2 may decrease the effectiveness of cisplatin, requiring higher doses of this highly toxic chemotherapeutic agent. The physiologic reason for this drug-drug interaction is currently unknown. Compound 3 is an indolealkylamine or tryptamine that, while having high affinity for the 5-HT1D receptor, also binds to other receptors within the 5-HT1 family, including 5-HT1B (originally classified as a 5-HT1D receptor subtype) and, to a lesser extent, 5-HT1A. It has no affinity for 5-HT2 or 5-HT3 receptors. Compound 3 acts as an agonist at 5-HT1D and 5-HT1B sites in the trigeminal nerve and meningeal arteries, respectively. Stimulation of these receptors leads to vasoconstriction that brings relief to migraine headache sufferers. This action is not selective for the CNS however, and the ability of 5-HT1B/1D agonists to constrict peripheral arterial vessels through their stimulation of 5-HT1D receptors in vascular smooth muscle means they are contraindicated in patients with hypertension and/or coronary artery disease. Compound 3 and its active secondary amine metabolite have a relatively short half-life of 3 hours. Apply the chemical understanding gained from the SAR analysis to this patient’s specific needs to make a therapeutic recommendation. Sr. MT is in need of a long-acting 5-HT3 receptor antagonist to guard against the acute and delayed emetogenic action of both drugs that will be included in her chemotherapy regimen. Compound 1 (palonosetron) would be an ideal agent to assist her in managing this highly distressful response to cisplatin and doxorubicin. Given IV 30 minutes prior to chemotherapy, palonosetron has been shown to control acute chemotherapy-associated nausea and vomiting, as well as the delayed CINV that can occur up to 5 days after drug administration. Days 2-3 are notoriously the worst for delayed CINV, and palonosetron shows a significantly greater antiemetic activity during this time period than other 5-HT3 antagonists. The impact of this drug on cardiovascular function is negligible, so it should not pose toxicity problems given Sr. MT’s underlying hypertension and hyperlipidemia. Compound 2 (ondansetron) would have a protective effect against acute CINV similar to palonosetron, but it does not have a duration of action sufficiently long to adequately manage the delayed nausea and vomiting that this patient will undoubtedly face. In addition, while not always observed in the clinic, it could potentially diminish the efficacy of the cisplatin, necessitating an increase in dose that could increase Sr. MT’s risk of cisplatin-induced ototoxicity and nephrotoxicity. Ondansetron would be very useful in some chemotherapy regimens, but not optimally beneficial in this one. Compound 3 (zolmitriptan) is the wrong drug to select for several reasons. First and foremost, it has no affinity for the 5-HT3 receptor and, therefore, cannot antagonize it to bring about the desired antiemetic effect. It does bind to 5-HT1 receptors, but acts as an agonist rather than an antagonist. While stimulating central 5-HT1A receptors could conceivably bring about some anxiolytic effects, the impact would be minimal since the affinity of zolmitriptan for this subtype is low. Finally, zolmitriptan’s predominant action in the periphery (5-HT1D receptor stimulation in vascular smooth muscle) would cause Sr. MT’s normally high blood pressure to rise to potentially dangerous levels. Victoria F. Roche, Ph.D. and S. William Zito, Ph.D.