Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Protein structure prediction wikipedia , lookup
List of types of proteins wikipedia , lookup
Homology modeling wikipedia , lookup
Protein folding wikipedia , lookup
Bimolecular fluorescence complementation wikipedia , lookup
Intrinsically disordered proteins wikipedia , lookup
Protein domain wikipedia , lookup
Protein mass spectrometry wikipedia , lookup
Protein purification wikipedia , lookup
Nuclear magnetic resonance spectroscopy of proteins wikipedia , lookup
Western blot wikipedia , lookup
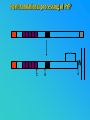
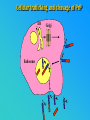
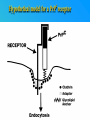
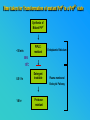
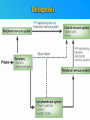
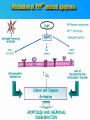
PRIONS THE INFECTIOUS PROTEINS Paras Yadav*, Jaspreet Singh Arora*, Sachinandan De*, Tirtha Kumar Datta*, Surender Lal Goswami*, Aarti Bhardwaj$, Shalini Jain# and Hariom Yadav# *Animal Biotechnology, #Animal Biochemistry Division, National Dairy Research Institute, Karnal-132001, Haryana, India $Meerut Institute of Engeenering and Technology, Meerut, U.P., India INTRODUCTION • Stanley B. Prusiner coined the term proin from Proteinaceous infective particle and changed to prion to sound it rhythmic. • Prion diseases were caused by misfolded proteins. • Elucidated the gene and mechanism by which wild type protein bring about the clinical disease. Prion Diseases Human • Kuru Animal • Scrapie • Fatal Familial Insomnia (FFI) • Bovine Spongiform Encephalopathy (BSE) • Creutzfeldt-Jakob disease (CJD) • Chronic Wasting Disease (CWD) Classification of prion diseases • Infectious/Exogenous – e.g., Kuru, BSE (mad cow disease), Scrapie – Spread by • Consumption of infected material. • Transfusion. • Sporadic • Familial/Hereditary – Due to autosomal dominant mutation of PrP. Differences between cellular and scrapie proteins PrPC PrPSC Solubility Soluble Non soluble Structure Alpha-helical Beta-sheeted Multimerisation state Monomeric Multimeric Infectivity Non infectious Infectious Susceptibility to Proteinase K Susceptible Resistant Steps in the biosynthesis of PrP c Post-translational processing of PrP S C B S A Cellular trafficking and cleavage of PrP ER Endosome Golgi Mechanism of Internalization of PrP C c Hypothetical model for a PrP receptor Model for the function of LRP- LR as the receptor for PrP BDII (aa 53-93) BDI (aa 144-179) PrP HSPG Dependent binding domain Direct binding domain (aa 161-179) LRP/LR GPI Heparan sulfate chain (HS) Proteogl ycan moiety Sulfated domains HSPG Cell Culture Systems for Prion Propagation Sequence of prion protein • Antioxidative Functions of Prion protein PrPC + Cu (Copper) Antioxidant activity Resistance to oxidative stress Prevent neuronal dysfunction (Brown et al., 2002) • Other functions c Models for the conversion of PrP to PrP sc c Sc Time taken for Transformation of mutant PrP to a PrP state Synthesis of Mutant PrPc PIPLCresistant <10 min Endoplasmic Reticulum BFA 180C 0.5-1 hr Detergent insoluble Plasma membrane/ Endocytic Pathway 1-6 hr Proteaseresistant Effect of conformation of PrP on Pro K Model of the cellular pathways Sc involved in generation of PrP c Proposed model of PrP aggregation and induction of CtmPrP Pathogenesis sc Mechanism of PrP induced apoptosis What are Calpains? They generate a C-terminal fragment(C2) which has molecular weight of 27-30 kDs. Increase in intracellular levels of Calcium increase production of terminal fragments . Calpastatin prevents production of C2. Inhibitors of lysosomal proteases has no effect on C2 production. Telling et al.,2004 Role of Caspases It was proposed that prion-associated toxicity involves altered trafficking of PrPc. Inhibition of ubiquitin-proteasome system(UPS). Deposition of aggresomes of PrPSc in nerve cells. Induction of Apoptosis with activation of Caspase 3 and Caspase 8. Complete molecular basis for neuronal death is not known. Aggregates of over expressed PrPc does not cause cell death. Tabrizi et al., 2005 Factors Responsible for Prion Propagation The AGAAAAGA Palindrome in Prion Generation Norstrom & Mastrianni, 2005 Factors Responsible for Prion Propagation cont… • PrPc association with lipid rafts in the early secretory pathway. • Creutzfeldt–Jakob disease (CJD) in Libyan Jews, linked to the E200K mutation in PRNP (E200KCJD). Model for chaperone-supervised PrP conversion E.g. Hsp70, GroEL Factors that prevent Prion Replication Phospholipase A2 Inhibitors prevent prion replication. Platelet-activating Factor Antagonists also inhibits prion replication. Bate et al.,2004 Drugs which share a N-benzylidene-benzohydrazide core structure. Bertsch et al.,2005 Trimethylamine N-oxide (TMAO), can prevent formation of PrPSc. Bennion, 2004 Gross and Microscopic Changes Gross changes Grossly there is Cortical atrophy and may also be present. ventricular dilatation Microscopic changes Scrapie BSE Kuru CJD Other microscopic changes • Gliosis within plaques. • Loss of oligodendrocytes within plaques. • Axons usually remain intact in plaques. • Both CD4+ and CD8+ lymphocytes are present in active lesions. (Kretzschmar et al.,1996, Wilesmith et al., 1997). Diagnosis 1. 2. 3. 4. 5. 6. Diagnosis can be made by: Clinical signs and Symptoms. Detection of Scrapie Associated fibrils. Detection of Abnormal Prion protein (PrPsc) by Western blotting. Two dimensional Gel Electrophoresis. Imunodiagnosis of Prion disease. Bioassay in Mice. Scrapie Associated fibrils. Mechanism of plaque formation PrPC PrPSC PrPC PrPsc fibrils Plaque Conclusion Molecular hallmark of the disorder is the accumulation of abnormal prion protein(PrPSc). Physiological functions of cellular prion protein (PrPc) is not clear. Identity of intracellular compartment where PrPc to PrPSc occurs is not established. Prion peptide of 106-126 residues is found to be neurotoxic. Studies on prion protein will open the avenues for treatment of other neurodegenerative disorders.