Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Behavioural genetics wikipedia , lookup
Genome (book) wikipedia , lookup
Frameshift mutation wikipedia , lookup
Microevolution wikipedia , lookup
Molecular Inversion Probe wikipedia , lookup
Population genetics wikipedia , lookup
Medical genetics wikipedia , lookup
Point mutation wikipedia , lookup
Sequence alignment wikipedia , lookup
Human genetic variation wikipedia , lookup
Multiple sequence alignment wikipedia , lookup
Epigenetics of neurodegenerative diseases wikipedia , lookup
Public health genomics wikipedia , lookup
Полиморфизм генома человека
Василий Раменский,
Институт молекулярной биологии
им. Энгельгардта РАН , Москва
Алма-Ата, 15.04.06
People are different…
…and so are their genomes
…caccagctcctgtgGggggaggccctgct…
…caccagctcctgtgGggggaggccctgct…
…caccagctcctgtgGggggaggccctgct…
…caccagctcctgtgCggggaggccctgct…
…caccagctcctgtgCggggaggccctgct…
Определение
SNP (single nucleotide polymorphism): существование в популяции
на одной и той же позиции геномной ДНК двух нуклеотидных
вариантов с частотой более редкого варианта (аллеля) ≥1%
5’---------------A---------------3’
|||||||||||||||||||||||||||||||
3’---------------T---------------5’
Na
5’---------------G---------------3’
|||||||||||||||||||||||||||||||
3’---------------C---------------5’
Ng
Na+Ng = N, Na/N ≥0.01, Ng/N ≥0.01
Комментарии к определению
•речь идет о сравнении последовательностей одного биол. вида
•слово «полиморфизм» не имеет в русском языке
множественного числа (Н.Ляпунова, личное сообщение)
•в обыденной речи под «полиморфизмом» чаще всего
подразумевают именно нуклеотид (т.е. используют его как
синоним слова «мутация»)
•определение подразумевает достоверное измерение частот в
популяции(-ях), что в текущей практике пока редкость
Типы полиморфизма в геноме
* однонуклеотидный (SNP)
* короткая вставка/делеция
* микросателлитный повтор различной длины (VNTR,
variable number tandem repeat)
* вставка объекта
* множественный нуклеотидный (MNP)
Некоторые свойства SNPs
• Comprise the ~90% of human genetic variation
• Occur with an average density ~1/600 bp
• Transition C↔T(G↔A) occurs at ~2/3 of all cases, three
transversions C↔A (G↔T), C↔G(G↔C), T↔A(A↔T) in
~1/6 of all cases each
• Most of them (~85%) are common to all populations
(with differing allele frequencies)
Why SNPs are important?
• Convenient genetic markers
• Responsible for existence of various phenotypes,
with primary interest in disease ones
• Pharmacogenomics: individual response to drugs
• Clues to understand human evolution
SNP в геноме человека
Классификация SNP по положению в геноме
1. гены
1.1 UTR
1.2 экзоны (cSNP)
1.2.1 синонимичные(sSNP)
1.2.2 несинонимичные (nsSNP)
1.3 интроны
1.4 сайты сплайсинга
2. регуляторные участки генов (rSNP)
3. межгенные участки
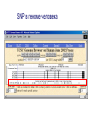
Synonymous vs. non-synonymous SNPs:
Example: Lysosomal alpha-glucosidase precursor (SwissProt P10253)
Hypothetical SNP: C T
HGVBase ID: SNP000003023 G C
…CAC CAG CTC CTG TGG GGG GAG GCC CTG CT…
…CAC CAG CTC CTG TGC GGG GAG GCT CTG CT…
… H
Q
L
L
W
G
E
A
L
…
… H
Q
L
L
C
G
E
A
L
…
nsSNP Trp746Cys
sSNP Ala749Ala
Summary of Annotation on human Genome Build 33
dbSNP Build 124 :
FUNCTION
CLASS
CODE
1
GENE
COUNT
SNP COUNT
338787
FUNCTIONAL
CLASSIFICATION
26210 Locus region
39214
Allele synonymous to contig
14342 nucleotide
4
50772
Allele nonsynonymous to contig
15710 nucleotide
5
546965
6
2925773
7
832
8
89554
9
7111
3
17898 untranslated region
19332 intron
769 splice site
18655 Allele is same as contig nucleotide
1006 Coding: synonymy unknown
Жизненный цикл SNP (по Miller&Kwok, 2001)
I.
Появление нового аллельного варианта путем мутации
(~100 мутаций на индивидуум)
II. «Выживание» до момента появления гомозигот по этому
аллелю
III. Медленное увеличение частоты в популяции
IV. Фиксация нового аллеля (0 vs. 100%), превращение в
between-species difference
Замечание
Описанный выше жизненный цикл SNP занимает ~0.3 млн
лет. Предполагая, что разделение человека и шимпанзе
произошло ~5 млн лет назад, а выход H.sapiens из Африки и
разделение различных популяций ~0.1-0.2 млн лет назад,
понятно отсутствие (а) одинаковых SNPs у человека и других
видов, (б) «private» SNP, т.е. локализованных в пределах
одной человеческой популяции
Why polymorphisms are maintained
in the population?
• Selectionists: because heterozygotes have
higher fitness
• Neutralists: because all observed
polymoprhisms are selectively neutral
- - - - - -- - - - - - - - - - - - - - - - - - - - - - - - Reality: is always somewhat more complicated
Why SNPs are important?
• Convenient genetic markers
• Responsible for existence of various phenotypes,
with primary interest in disease ones
• Pharmacogenomics: individual response to drugs
• Clues to understand human evolution
nsSNPs vs. disease mutations
Disease mutations are rare (<<1%) and usually cause
monogenic diseases (e.g., cystic fibrosis)
nsSNPs are frequent (>1%) and can modify risks of
major common (multigenic, complex) diseases (e.g.,
cancer, cardiovascular disease, mental illness,
autoimmune states, diabetes)
In some cases, however, it is difficult to make a distinction
Some common nsSNPs are known to affect
critical structure features
Frequency of the haemochromatosis allelic variant of
HLA-H protein Cys260Tyr (with destroyed disulphide
bond) is up to 6% in Northern Europe
Application area for prediction methods
Genetics of complex diseases
Analysis of human birth defects
Genetics of rare developmental phenotypes (analysis of
de novo mutations that cannot be mapped by genetic
techniques)
Genetics of model organisms (identification of genes
involved in diverse processes by mutagenesis screens)
Genomics and evolutionary genetics (e.g., quantifying
selective pressure)
Identifying SNPs responsible for
complex diseases: general strategies
whole genome scan – hypothesis free
approach; extraordinary number of candidate SNPs
candidate gene studies – requires a priori
models; nevertheless, large numbers of candidate
SNPs must be tested
Identifying SNPs responsible for
complex diseases: application
1. A SNP with established association need not be
functional; therefore, in silico expertise is required
for selection of potentially functional SNPs
2. Detection of enrichment of rare potentially
functional alleles in the disease population (plasma
levels of HDL-cholesterol, hypertension, colorectal
cancer)
Methods for prediction of effect of nsSNPs
* Sequence-based methods: analysis of multiple
alignment with homologs Ng-Henikoff [2002]
* Structure-based methods: analysis of various
structural parameters Wang, Moult [2001]; Chasman, Adams [2001]
* Combined methods: sequence and structure analysis
Sunyaev,Ramensky,Bork [2000, 2001, 2002]
PolyPhen: prediction of amino acid
substitution effect on protein function
Prediction: benign (neutral), damaging (deleterious)
PolyPhen: prediction of amino acid
substitution effect on protein function
Data sources:
1. Sequence annotation of the query protein
2. PSIC profile matrix values derived from multiple
alignment with homologous proteins
3. Structural parameters and contacts of query protein
structure or its >50% homolog
Prediction: benign (neutral), damaging (deleterious)
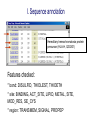
I. Sequence annotation
Hereditary hemochromatosis protein
precursor (HLA-H, Q30201)
Features checked:
* bond: DISULFID, THIOLEST, THIOETH
* site: BINDING, ACT_SITE, LIPID, METAL, SITE,
MOD_RES, SE_CYS
* region: TRANSMEM, SIGNAL, PROPEP
II. PSIC: profile analysis of
homologous sequences
1. Align with homologous proteins with seq. ide. 30..94%
II. PSIC: profile analysis of
homologous sequences
2. Calculate the profile matrix with PSIC algorithm
Profile matrix: Sa,j = ln[ pa,j / qa ], a = {1,..20}, j = {1,..N}, N =
alignment length
SAsn,4
SCys,4
II. PSIC: profile analysis of
homologous sequences
3. Analyse difference between profile scores for two a.a.
variants:
AsnCys: = | SAsn,4 – SCys,4 | = 1.591
SAsn,4
SCys,4
III. 3D structure analysis
1. Residues that are in spatial contact with a
ligand or other “critical” residues
Zen 999
residues in 5Å contact
with Zen 999
Bos Taurus trypsin
[PDB ID :1ql7]
III. 3D structure analysis
2. Residues that form the hydrophobic core of
the protein (buried residues)
Surface residues
Buried residues
Bos Taurus trypsin
[PDB ID :1ql7]
Structural parameters and contacts
Secondary structure
Phi-psi dihedral angles
Solvent accessible surface area, normed s.a.s.a
Change in accessible surface propensity
Change in residue side chain volume
Contacts with heteroatoms
Interchain contacts
Contacts with functional sites (BINDING,
ACT_SITE, LIPID, and METAL)
Region of the phi-psi map (Ramachandran map)
Normalised B-factor (temperature factor)
RULES (connected with logical AND)
PREDICTION
PSIC score
difference :
Substitution site properties:
arbitrary
annotated as a functional* or bond formation**
site
arbitrary
in a region annotated or predicted as
transmembrane
PHAT matrix difference resulting
from substitution is negative
0.5
arbitrary
arbitrary
benign
>1.0
atoms are closer than 3.0Å to atoms of a ligand
or residue annotated as BINDING, ACT_SITE,
LIPID, METAL
arbitrary
probably damaging
not considered
normed accessibility ACC15%
0.5<1.5
normed accessibility ACC5%
1.5<2.0
>2.0
Substitution type properties:
absolute change of accessible
surface propensity is 0.75 or
absolute change of side chain
volume is 60
absolute change of accessible
surface propensity is 1.0 or
absolute change of side chain
volume is 80
probably damaging
possibly damaging
possibly damaging
probably damaging
arbitrary
arbitrary
possibly damaging
arbitrary
arbitrary
probably damaging
Validation: control sets
all
dam unknown dam/(dam+ben)
–––––––––––––––––––––––––––––––––––––––––––––
Disease mutations
Strict set
444 366 3
82.9%
Total
2,782 2,047 70
75.4%
Between species substitutions
Total
671 58
5
8.7%
Validation: case studies
• APEX1 protein: 24 out of 26 substitutions predicted
correctly (Xi et al.)
• Plasminogen activator inhibitor-2: 18 out of 20 (Di
Guisto et al.)
• 3 HapMap populations and 10 primate species:
analysis of ~27,000 nsSNPs with frequencies (Victoria
Carlton, AFFYMETRIX, private communication)
Validation: allele frequency
Validation: nsSNPs vs. human-mouse
interspecies variation
PolyPhen predictions for dbSNP b.121
[ Ivan Adzhubei, 2004 ]
All:
9,502
27,991
7,905
5,521
50,919
unknown
benign...............67.6%
possibly damaging....19.1%
probably damaging....13.3%
total (44,005 unique rs’s)
With structure:
42
2,142
531
1,076
3,791
unknown
benign...............57.1%
possibly damaging....14.2%
probably damaging....28.7%
total (,167 uniqe rs’s)
PolyPhen predictions for dbSNP b.121
[ Ivan Adzhubei, 2004 ]
All:
Filtered: 5 seq. in multiple alignment
16,813
5,195
4,168
26,176
benign...............64.2%
possibly damaging....19.8%
probably damaging....15.9%
total (21,677 unique rs’s)
With structure:
Filtered: 5 seq. in multiple alignment
2,021
499
1,050
3,570
benign...............56.6%
possibly damaging....14.0%
probably damaging....29.4%
total (2,983 unique rs’s)
Hydrophobic core stability parameters
are the best predictors
Ramensky et al., Nucleic Acids Res. (2002) 30:3894-90
PolyPhen http://www.bork.embl.de/PolyPhen
PolyPhen input :
Protein identifier
OR sequence
Substitution
position
Substitution type
PolyPhen http://www.bork.embl.de/PolyPhen
PolyPhen:
nsSNPs data collection
Transphyretin
(PDB: 1tyr,
SNP000012365)
Thr118 Asn occurs
at the ligand (REA)
binding site
Thr 118
REA 130
DAMAGING nsSNPs
Trypsin
(PDB: 1trn,
SNP000012965)
Ser142Phe results
in the strong side
chain volume change
at a buried position
DAMAGING nsSNPs
Ser 142
Damaging nsSNPs
• We estimate that ~20% of non-synonymous cSNPs
from databases are damaging
• Average allele frequency of non-synonymous cSNPs
predicted to be damaging is twice lower than for
benign non-synonymous cSNPs
• We propose to use these predictions for prioritisation
of candidates for association studies
Development directions
• Better multiple alignment pipeline
• Compensated nsSNPs
• Non-globular structural regions
• Non-coding SNPs
An example of compensated
pathogenic deviation
Polyphenism: the ability of a single genome to produce two or more
alternative morphologies within a single population in response to an
environmental cue (such as temperature, photoperiod, or nutrition).
[Dr. Ehab Abouheif, McGill University, Montréal Québec]
The seasonal morphs of the buckeye butterfly, Precis coenia (Nymphalidae). The
ventral surfaces are shown. The Summer morph ("linea") is on the left; the Fall morph
("rosa") is on the right. [Scott F.Gilbert, A Companion to Developmental Biology.
Chapter 22, Seasonal Polyphenism in Butterfly Wings]
People
Shamil Sunyaev(1), Vasily Ramensky(2), Steffen Schmidt(1), Ivan Adzhubei(1)
(1) Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard
Medical School, Boston, USA) (2) Engelhardt Institute of Molecular Biology Moscow Russia)
Peer Bork, Yan P. Yuan (European Molecular
Biology Laboratory, Heidelberg, Germany)























































![[edit] Use and importance of SNPs](http://s1.studyres.com/store/data/004266468_1-7f13e1f299772c229e6da154ec2770fe-150x150.png)