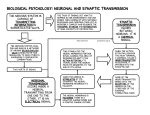
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Electrophysiology wikipedia , lookup
Nervous system network models wikipedia , lookup
Multielectrode array wikipedia , lookup
Stimulus (physiology) wikipedia , lookup
Optogenetics wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Biological neuron model wikipedia , lookup
Feature detection (nervous system) wikipedia , lookup
Subventricular zone wikipedia , lookup
Development of the nervous system wikipedia , lookup
NeuroToxicology 31 (2010) 277–290 Contents lists available at ScienceDirect NeuroToxicology Quantitative assessment of neurite outgrowth in human embryonic stem cell-derived hN2TM cells using automated high-content image analysis§ Joshua A. Harrill a, Theresa M. Freudenrich a, Dave W. Machacek b, Steven L. Stice b,c, William R. Mundy a,* a Systems Biology Branch, Integrated Systems Toxicology Division, National Health and Environmental Effects Research Laboratory, United States Environmental Protection Agency, Research Triangle Park, NC 27711, United States b ArunA Biomedical, Athens, GA 30602, United States c Regenerative Bioscience Center, University of Georgia, Athens, GA 30602, United States A R T I C L E I N F O A B S T R A C T Article history: Received 14 December 2009 Accepted 17 February 2010 Available online 25 February 2010 Throughout development neurons undergo a number of morphological changes including neurite outgrowth from the cell body. Exposure to neurotoxic chemicals that interfere with this process may result in permanent deficits in nervous system function. Traditionally, rodent primary neural cultures and immortalized human and non-human clonal cell lines have been used to investigate the molecular mechanisms controlling neurite outgrowth and examine chemical effects on this process. The present study characterizes the molecular phenotype of hN2TM human embryonic stem cell (hESC)-derived neural cells and uses automated high-content image analysis to measure neurite outgrowth in vitro. At 24 h post-plating hN2TM cells express a number of protein markers indicative of a neuronal phenotype, including: nestin, bIII-tubulin, microtubule-associated protein 2 (MAP2) and phosphorylated neurofilaments. Neurite outgrowth in hN2TM cells proceeded rapidly, with a majority of cells extending one to three neurites by 48 h in culture. In addition, concentration-dependent decreases in neurite outgrowth and ATP-content were observed following treatment of hN2TM cells with either bisindolylmaleimide I, U0126, lithium chloride, sodium orthovanadate and brefeldin A, all of which have previously been shown to inhibit neurite outgrowth in primary rodent neural cultures. Overall, the molecular phenotype, rate of neurite outgrowth and sensitivity of hN2TM cells to neurite outgrowth inhibitors were comparable to other in vitro models previously characterized in the literature. hN2TM cells provide a model in which to investigate chemical effects on neurite outgrowth in a non-transformed human-derived cells and provide an alternative to the use of primary rodent neural cultures or immortalized clonal cell lines. Published by Elsevier Inc. Keywords: Neurite outgrowth High-content analysis Human embryonic stem cell-derived neural culture 1. Introduction During the differentiation of precursor cells to a committed neuronal lineage, newly formed neurons undergo a series of extensive morphological changes as they mature including emergence of neurites, neurite outgrowth, neurite branching and establishment of cell–cell contacts (i.e. synaptogenesis). These morphological changes are necessary, although not sufficient, for the formation of the intricate network of neural circuits that facilitate nervous system function (Sanes et al., § This manuscript has been reviewed by the National Health and Environmental Effects Research Laboratory, U.S. Environmental Protection Agency, and approved for publication. Approval does not signify that the contents reflect the views of the Agency, nor does mention of trade names or commercial products constitute endorsement or recommendation for use. * Corresponding author at: USEPA, Integrated Systems Toxicology Division, B10506, Research Triangle Park, NC 27711, USA. Tel.: +1 919 541 7726. E-mail address: [email protected] (W.R. Mundy). 0161-813X/$ – see front matter . Published by Elsevier Inc. doi:10.1016/j.neuro.2010.02.003 2006). Early pre- and post-natal exposure to neurotoxic compounds can interfere with these developmental events and could potentially result in deficits in nervous system function in later life stages (Rice and Barone, 2000; Costa et al., 2004; Grandjean and Landrigan, 2006). Neurite outgrowth, a critical component of this developmental chain of events, can be recapitulated in vitro using a variety of cell models, such as nervous system derived clonal cell lines and primary neural cultures from the mammalian CNS. These models have become valuable tools for studying the molecular mechanisms that control neurite outgrowth (Zhang et al., 2009a,b; Yu and Malenka, 2003; Redmond et al., 2002; Jin et al., 2003; Khaibullina et al., 2004) and for investigating the mechanism(s)-of-action for known developmental neurotoxicants (Yamauchi et al., 2007; Lein et al., 2000; Howard et al., 2005; Audesirk et al., 1991). It has also been proposed that in vitro measures of neurite outgrowth can be useful in high-throughput screening assays (Radio et al., 2008, 2010) as a means to identify potential developmental neurotoxicants (Radio and Mundy, 2008). 278 J.A. Harrill et al. / NeuroToxicology 31 (2010) 277–290 A number of recent publications advocate the use of in vitro cell culture models as tools for efficient identification and prioritization of chemicals that may be hazardous to humans (NRC, 2007; Coecke et al., 2007; Lein et al., 2005). Specifically, a report by the National Academy of Sciences entitled ‘Toxicity testing in the 21st century: a vision and a strategy’ emphasizes the use of in vitro models derived from human tissues (NRC, 2007). The impetus for this point-of-view being that use of in vitro toxicity assays in human-derived cells, as opposed to cells from non-human mammalian species, may decrease some of the uncertainties involved in evaluating the effects of chemicals and applying that knowledge to address human risk (i.e. interspecies extrapolation) (NRC, 2007). In the case of assessing neurite outgrowth, there are a number of immortalized and tumor-derived neural cell lines of human origin currently available (Radio and Mundy, 2008; Harry and Tiffany-Castiglioni, 2005) as well as reliable methods for the culture of primary rodent neurons (Higgins and Banker, 1998). However, transformed clonal cell lines or rodent primary neural cultures may not accurately represent human nervous system biology (LePage et al., 2005; Allen et al., 2005). The response or sensitivity of neural cultures to toxic compounds may differ across species or across models as noted in previous reports examining the effects of ethanol, staurosporine and mercury on the processes of neural development in vitro (Breier et al., 2009; Moors et al., 2009; Cedrola et al., 2003). In the context of evaluating chemicals as potential developmental neurotoxicants in humans, a human stem cell-derived culture model may prove more informative than transformed cell lines and also circumvent problems associated with primary human neural cell availability (McNeish, 2004). Recent advances in stem cell biology have resulted in methods in which embryonic stem cells of human origin can be differentiated along a neuronal lineage and grown in dissociated cultures (Reubinoff et al., 2001; Zhang et al., 2001). Given the proper extracellular cues and growth substrate, the maturing cells can display morphological characteristics and express a number of protein markers indicative of a neuronal lineage (Reubinoff et al., 2001; Zhang et al., 2001; Shin et al., 2006). These stem cell-derived models may serve as valuable tools for examining chemical effects on neuronal maturation, including neurite outgrowth, using human cells. In the context of high- to medium-throughput chemical screening, the use of stem cell-derived neuronal cultures also has some potential caveats. Namely, the time- and laborintensive process of differentiating a proliferative population of stem cells (hESC) to a population of terminally differentiated neurons, which can take weeks (Reubinoff et al., 2001; Zhang et al., 2001; Shin et al., 2005). The present study describes the phenotypic characteristics and measures neurite outgrowth in hN2TM cells, a novel, commercially available, hESC-derived neuronal model which is provided in a pre-differentiated state for rapid end user applications (ArunA Biomedical, Athens, GA). The hN2TM cell line is derived from neuroepithelial cells of WA09 hESC (Thomson et al., 1998) origin according to a previously described protocol (Shin et al., 2005, 2006). Importantly, as opposed to other methods of deriving neural progenitors through three-dimensional neurosphere and embryoid body formations (Reubinoff et al., 2001; Zhang et al., 2001), these adherent monolayer cultures are uniformly exposed to growth factors and/or morphogens throughout their propagation. Prior to differentiation into hN2TM cells the population was confirmed karyotypically normal, >95% nestin positive and <3% OCT-4 positive (Shin et al., 2006). The cells were produced in bulk by propagation for an additional 2 weeks beyond the neuroepithelial stage by removal of bFGF from the media and cryopreserved (ArunA Biomedical, Athens, GA) for end user applications. In the present study, the utility of dissociated hN2TM cultures as an in vitro model for neurite outgrowth was assessed using automated high-content image analysis (HCA). In addition, the molecular phenotype of these cells was examined using immunocytochemical staining. 2. Methods 2.1. Materials hN2TM human neural cells, growth media and supplements were obtained from ArunA Biomedical, Inc. (Athens, GA). The growth substrates poly-L-lysine and laminin were purchased from Sigma–Aldrich (St. Louis, MO). Bisindolylmaleimide I (Bis1) and brefeldin A were purchased from Calbiochem, Inc. (San Diego, CA). Dimethyl sulfoxide (DMSO, dosing vehicle), lithium chloride (LiCl) and sodium orthovanadate (Na3VO4) were purchased from Sigma– Aldrich (St. Louis, MO). U0126 was purchased from Promega Corp. (Madison, WI). Hoechst 33258 dye, immunocytochemical staining buffer (ISB), mouse monoclonal antibody against bIII-tubulin and DyLight1 488-conjugated rabbit anti-mouse IgG secondary antibody were components of a Cellomics1 Neurite Outgrowth HitKitTM purchased from ThermoFisher Scientific, Inc. (Waltham, MA). Mouse monoclonal antibodies for microtubule-associated protein 2 (MAP2) and nestin were purchased from Millipore, Inc. (Billerica, MA). Mouse monoclonal antibody SMI-312 which detects phosphorylated forms of a variety of axonal neurofilaments was purchased from Covance, Inc. (Princeton, NJ). 2.2. Cell culture Costar1 96-well polystyrene cell culture dishes (Corning, Inc., Corning, NY) were coated with a solution of 50 mg/ml poly-L-lysine in sterile H2O for 2 h (37 8C), rinsed once with sterile H2O and then coated with a solution of 20 mg/ml laminin in sterile phosphatebuffered saline (PBS) for 2 h. Plates were then rinsed once with warm PBS prior to plating of hN2TM cells. Cells were stored at 70 8C and thawed at time of use. After thawing at 37 8C, cells were suspended in serum-free ArunA basal medium supplemented with ArunA Neural Supplement (ANSTM), leukemia inhibitory factor (LIF, 10 ng/ml), penicillin (50 U/ml), streptomycin (50 mg/ml) and 2 mM L-glutamine. A small aliquot of cells were then stained with 0.4% trypan blue and counted on a hemocytometer. Live cell yields post-thawing ranged from 60 to 80%. Cells were plated at densities ranging from 2500 to 10,000 cells/well (8.33 103 to 3.33 104 cells/cm2, respectively) based on the number of live cells counted. The number of cells per cm2 (i.e. plating density) was calculated by dividing the number of cells per well by the well area (0.3 cm2). Cells were maintained in a humidified incubator at 37 8C with a 95% air/5% CO2 atmosphere. 2.3. Chemical treatment Concentration ranges for the five test compounds were as follows: brefeldin A (0.01, 0.03, 0.1, 0.3, 1 mM), Bis1 (0.1, 0.3, 1, 3, 10 mM), U0126 (0.3, 1, 3, 10, 30 mM), sodium orthovanadate (1, 3, 10, 30, 100 mM) and lithium chloride (0.3, 1, 3, 10, 30 mM). Guidance for concentration range selection was based on previously published works cited in Table 1. Stock solutions (1000) of the highest tested concentration of Bis1, brefeldin A and U0126 were prepared in pure DMSO and stock solutions for the remainder of the concentration ranges were prepared by serial dilution in DMSO. Dosing solutions for each chemical concentration were prepared by diluting stock solutions 1:100 in ArunA basal media. Stock and dosing solutions of LiCl and Na3VO4 were prepared using the same method, save that stock solutions were prepared in ArunA basal medium as opposed to DMSO. 10 ml of dosing solutions were then added to the cell culture wells J.A. Harrill et al. / NeuroToxicology 31 (2010) 277–290 279 Table 1 Chemicals with evidence of neurite outgrowth inhibition in rodent primary neural cultures. Compound Study Cell type a,b Concentration range Effects Bis1 U0126 Radio et al. Radio et al. CGC (PND7) CGC (PND7) 1 nM–100 mM 1 nM–100 mM # in neurite outgrowth # in neurite outgrowth LiCl Takahashi et al. Munoz-Montano et al. Hollander and Bennett Hippocampal (E18)c CGC (PND7) DRG (E8-9)d 2–15 mM 1–20 mM 25 mM # ratio of axon length to cell body diameter Biphasic " then # in % cells with long neurites # in neurite length Na3VO4 Bref A Mandel and Banker Jareb and Banker Hippocampal (E18) Hippocampal (E18) 25–100 mM 0.14–3.57 mM # length of longest neurite # in % of neurons with axons a b c d PNDx = post-natal day x. CGC = cerebellar granule cells. Ex = embryonic day x; designates the age of mouse or rat pups at the time of culture. DRG = dorsal root ganglia. containing growing hN2TM cells in 90 ml of media to achieve the nominal media concentrations listed above. Final DMSO concentrations were 0.1% for all treatment wells containing varying concentrations of brefeldin A, Bis1, U0126 and corresponding vehicle control wells. Dose solutions were applied to the cells 2 h after plating. Cells were then returned to the incubator. ATPcontent and neuronal morphology were then examined at either 2, 6, 24 or 48 h as detailed below. The effects of DMSO on ATP-content and neuronal morphology were examined by preparing dosing solutions of 0.5–50% DMSO in ArunA basal media and applying to tissue culture wells at a 1:10 dilution (final concentration range examined: 0.05–5% DMSO). 2.4. Immunocytochemistry Cell cultures containing 100 ml of media volume were removed from the incubator and fixed in situ with 100 ml of a warm (37 8C) solution of 8% paraformaldehyde (PFA)/8% sucrose and 0.1% Hoechst 33258 dye in PBS for 20 min. This fixation method effectively preserved the fine morphological features of the cultures. Fixative was then gently aspirated and cells washed three times with immunocytochemical staining buffer (ISB). Primary antibodies diluted in ISB were then applied as follows: bIII-tubulin (1:800), MAP2 (1:800), nestin (1:400) and panaxonal neurofilament SMI-312 (1:200) for 1 h at room temperature. The entire antibody panel was used to characterize the neuronal phenotype of the hN2TM cells, while bIII-tubulin was specifically used to label cell bodies and neurites for high-content image analysis (HCA). Following incubation in primary antibodies, cells were washed three times with ISB and incubated with a 1:500 dilution of DyLight1 488-conjugated rabbit anti-mouse IgG secondary antibody in ISB for 1 h at room temperature, protected from light. Cells were then washed twice with ISB, twice with Dulbecco’s phosphate-buffered saline (PBS), and stored at 4 8C prior to image acquisition and analysis. 2.5. Measurements of hN2TM morphology bIII-Tubulin stained cell cultures were allowed to warm to room temperature. Plates were then loaded into a Cellomics ArrayScan VTI HCS reader high-content imaging system (ThermoFisher Scientific, Waltham, MA) for automated image acquisition and morphometric analyses. This system consists of an epifluorescent microscope with an EXFO X-citeTM 120 metal-halide arc lamp, motorized imaging objectives, stage and excitation/emission filter wheel and a 12-bit high-resolution CCD camera connected to a Dell Intel1 XenonTM computer terminal with 2 GHz processor. Image acquisition and storage was performed using the vHCS Scan software package, version 6.6.1.4. Matched fluorescent images of Hoechst-stained nuclei (Fig. 1A) and bIII-tubulin/DyLight1 488 immunolabeled cells (Fig. 1B) were acquired using 365/515 (channel 1) and 475/515 (channel 2) nm excitation/emission filter couplings, respectively, with a 20 objective (Zeiss, Inc., Thornwood, NY). Fixed integration times for image acquisition in each channel were determined by manual sampling of control-treated wells across multiple plates. A matching pseudocolored composite image of Hoechst-stained nuclei (blue) and bIII-tubulin/DyLight1 488 labeled cell bodies and neurites (green) is shown in Fig. 1C. Image analysis was performed in real-time with a manually optimized version of the Cellomics Neural Profiling Bioapplication v3.5. Optimization of nucleus and cell body selection criteria, as well as cell body masking and neurite tracing parameters, were determined a priori by using representative images from untreated cultures following 24 h of growth at a density of 7500 cells per well. Manual comparison of representative images from untreated control wells to matched tracing overlays was performed during optimization to insure the algorithm settings provided an accurate trace. A full listing of parameters for the algorithm used herein is available from the authors upon request. The Neural Profiling Bioapplication performs automated image analysis in a sequential manner as follows. Briefly, nuclei were identified in channel 1 as bright objects on a dark background (Fig. 1D). Nuclei with size and intensity values outside of the ranges determined a priori for viable cells were identified in the channel 1 image and rejected from further analyses (Fig. 1D, objects circled in orange). Spatial coordinates from the channel 1 image were then superimposed on the matching channel 2 image. Cell body masks in channel 2 were then cast based on positional data from channel 1 nuclei and a set of user-defined geometric and signal intensity-based parameters (Fig. 1E, blue and red traces). Cell bodies corresponding to valid neurons were then selected (Fig. 1E, blue traces) and invalid cell bodies rejected (Fig. 1E, red traces). Parameters for valid cell body selection include the presence of exactly one nucleus within the cell body mask, a requirement that the nucleus met the gating criteria imposed in channel 1, a requirement that at least 25% of the nucleus perimeter is bounded by DyLight1 488 labeled cytoplasm and a requirement that the total cell body area not exceed 4000 mm2. Neurites emerging from the selected cell bodies were then individually traced and measured (Fig. 1F). For this study, neurites were defined as processes >10 mm in length. Neurites were separated from cell bodies at points when the half-width of the labeled cytoplasm was less 3.6 mm across. In the case of neurites with an ambiguous origin (i.e. appearing to emerge from or contact multiple cell bodies) the Neural Profiling Bioapplication traced the neurite from all potential origin points and retained the longest neurite for measurements of length and number of neurites per neuron. This effectively prevented repeated sampling of the same neurite segment within each image. Morphometric data from high-content image analysis (HCA) included measurements of the average number of neurites per neuron and total neurite length per neuron. Data for both 280 J.A. Harrill et al. / NeuroToxicology 31 (2010) 277–290 Fig. 1. Automated measurement of neurite outgrowth in hN2TM cells. Cells were grown in 96-well titer plates at a density of 7500 cells/well (2.5 104 cells/cm2) for 24 h, fixed and fluorescently labeled. Cells were then imaged and neurite outgrowth measured. Images in panels A–F are 20 magnification high-resolution images obtained using the ArrayScan VTI. (A) Nuclei labeled with Hoechst 33258 and visualized in channel 1. (B) Cell bodies and neurites labeled with bIII-tubulin/DyLight1 488 in channel 2. (C) Pseudocolored composite image. (D) Nuclei are identified as bright objects on a dark field and masked; blue trace = selected nuclei, orange trace = rejected nuclei. An expanded view of nuclei bounded by the yellow box is given in the panel D inset to better illustrate nuclei traces. (E) Cell body masks based on fluorescent intensity of bIIItubulin/DyLight1 488 labeling and position of channel 1 nuclei; blue trace = accepted, red trace = rejected cell body. Yellow arrows denote cells with cell body size and shape parameters inside the accepted range for valid cells but are rejected due to a rejected nucleus in channel 1. Rejected nuclei with no discernable cell body do not generate a mask in channel 2 tracing. These cells are not included in the final measurements of average number of neurites per neuron or total neurite length per neuron. (F) Neurites emerging from accepted cell bodies are traced (light blue, green and purple lines) and quantified. Scale bars = 50 mm in all panels. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.) endpoints were collected on cell-by-cell basis. The number of neurites and the cumulative length of all neurites associated with each cell body (i.e. total neurite length) were calculated for each cell meeting the selection criteria outlined above. Cell-level measurements were then averaged to obtain a mean measurement for the average number of neurites per neuron and total neurite length per neuron for the cell populations sampled within each well. These well-level averages are reported in the present study and were treated as the statistical unit for analysis of neuronal morphology. In addition, the average number of neurons per field was measured as an indicator of cell health in treated cultures. Only cells that met the criteria for valid cell body selection, as listed above, are included in this measure of neuron density. Given that a uniform number of viable cells are plated in each well at the J.A. Harrill et al. / NeuroToxicology 31 (2010) 277–290 281 initiation of the cultures, a relative decrease in neuron density is interpreted as a decrease in cell health in the present concentration-response experiments. At 20 magnification, Cellomics Arrayscan VTI can sample 81 unique fields of view and quantify the number of neurons sampled per well in real-time. For time course, cell-density gradient and concentration-response studies, a sufficient number of fields were sampled so that at least 350 neurons were measured within each well. This was sufficient to minimize the variation observed in average morphometric measurements across wells (data not shown). 2.6. ATP-content ATP-content in chemical treated cultures was measured using a CellTiter-Glo1 Luminescent Cell Viability Assay Kit (Promega, Madison, WI). This assay uses a luciferase-catalyzed reaction to measure the level of adenosine 50 -triphosphate (ATP) in each well, which is produced by metabolically active cells. Radio et al. (2008) demonstrate that the amount of ATP present is proportional to the number of living cells in a well. Briefly, 100 ml of luminescent reagent was added to each well 22 h after exposure (i.e. 24 h after plating). Plates were gently mixed on an orbital shaker and stored, protected from light, for 30 min at room temperature. Luminescent signal was then quantified using a FLUOstar Optima plate reader (BMG LABTECH, Durham, NC). 2.7. Statistics In experiments measuring basal neurite outgrowth over time or evaluating the effects of plating density on neurite outgrowth, raw values from morphological measurements were analyzed. All time course and plating density experiments were performed twice using independent cultures with n = 4–6 wells per condition per culture. For concentration-response experiments, neurite outgrowth data were normalized within experiment to corresponding control wells prior to statistical analysis. In experiments with nonorganic molecules (LiCl, Na3VO4) or DMSO alone, data were normalized to untreated control wells. In experiments with organic compounds prepared in DMSO (Bis1, U0126, brefeldin A) data were normalized to vehicle control wells. In ATP-content experiments, luminescent signals were normalized to appropriate controls within each plate and analyzed across experiments. For each concentration-response examined, experiments were repeated two to three times using independent cultures as described in figure captions. Neurite outgrowth data and ATPcontent data were analyzed using a one-way ANOVA with a significance threshold of p < 0.05. This was followed by a Dunnett’s post hoc mean contrast test (p < 0.01) to determine if treatment group means were significantly different from corresponding control means, as described. Neurite outgrowth, neuron density and ATP-content data are presented as % change from control. Raw mean values standard deviations for neurite outgrowth measurements are provided throughout the text. Statistical analysis was performed using Graphpad Prism1 v5 (La Jolla, CA). 3. Results 3.1. Characterization of hN2TM cells Neurite outgrowth in hN2TM cells progresses rapidly following plating on polylysine and laminin coated 96-well plates. At the time of plating the cells appear spherical in shape with no apparent neurite growth. Within the first 2 h after plating, thin neurites begin to emerge from the cell body of a small proportion of cells (Fig. 2A). By 6 h many cells have neurites that are longer than the Fig. 2. Growth of hN2TM cells over time. Images of live cells grown for (A) 2, (B) 6 or (C) 24 h at a density of 7500 cells/well (2.5 104 cells/cm2). Images were taken at 20 magnification on a Nikon Eclipse TE200 microscope equipped with Hoffman modulation contrast optics. The arrow in panel (A) points to an emerging neurite. Scale bars = 100 mm. 282 J.A. Harrill et al. / NeuroToxicology 31 (2010) 277–290 broadest diameter of their respective cell bodies (Fig. 2B) and at 24 h a majority of viable cells have between one and three long neurites (Fig. 2C). At 24 h post-plating, hN2TM cells are positive for a number of protein markers indicative of a neuronal phenotype. The neuronal microtubule protein bIII-tubulin was present in both the cell bodies and all neurites of viable cells (Fig. 3A). Similarly, cell bodies and neurites were positive for nestin, an intermediate filament protein expressed in progenitor and newly differentiated cells of the neural epithelial lineage (Wiese et al., 2004) (Fig. 3B). In addition, cells grown for 24 h were immunolabeled with the SMI-312 primary antibody, which is targeted against phosphorylated neurofilament proteins and specifically labels axons in human fetal tissue (Ulfig et al., 1998). The resulting immunofluorescent signal was prominent in the long, thin processes emerging from the cell body of the cells (Fig. 3C). Less intense immunolabeling was also apparent within the cell body, but only at the points where neurites are emerging. Some thin processes not immunolabeled with SMI-312 were also observed upon comparison of fluorescent images to matching differential interference contrast images (Fig. 3C and G, arrows). At 24 h, the cells were also positive for the expression of MAP2 (Fig. 3D), a neuronal microtubule-associated protein enriched in dendrites (Caceres et al., 1986). Similar to the SMI-312 axonal marker, several neurites were observed that were not immunolabeled with the MAP2 antibody (Fig. 3D and H). For studies of neurite outgrowth in hN2TM cells using automated high-content microscopy, the bIII-tubulin primary antibody was used as it indiscriminately labeled all neurites, as well as cell bodies. 3.2. Measurement of neurite outgrowth in hN2TM cells using HCA Similar to the qualitative observations made during live cell imaging, automated measurement of hN2TM cells demonstrated a rapid increase in neurite outgrowth during the first 48 h after plating (Fig. 4). During this early growth phase, the average number of neurites per neuron increased significantly (Fig. 4A, 0.15 0.08 at 2 h; 1.28 0.07 at 24 h). The population distribution for this endpoint indicated that only 8.6% of cells had at least 1 neurite at 2 h, whereas >60% of cells had at least 1 neurite at 24 h (Fig. 4B). Very few cells (<2.5%) developed more than three primary neurites during the time period sampled (Fig. 4B). There was no significant difference in the average number of neurites per neuron between 24 and 48 h. Total neurite length per neuron significantly increased during the initial 48 h growth period (Fig. 4C). Total neurite length per neuron increased by more than 10-fold between 2 and 24 h (3.12 2.14 mm at 2 h; 56.5 2.3 mm at 24 h). Unlike the average number of neurites per neuron, neurite total length per neuron continued to increase after 24 h (73.10 7.9 mm at 48 h). For neurite outgrowth and ATP-content concentration-response experiments, cells were fixed and sampled at 24 h. At this time point, measurements of total neurite length had not yet reached an asymptote. This provided a dynamic range in which both chemically induced increases and decreases in neurite outgrowth could be detected. Changing the plating density of hN2TM cells between 2500 and 10,000 cells/well did not significantly affect neurite outgrowth measurements (Fig. 4D–F) at 24 h. For neurite outgrowth and ATPcontent concentration-response experiments, an intermediate cell number of 7500 cells/well was used. This cell number provided enough separation of cells along the plating surface for resolution of individual neurites with a 20 imaging objective. At 24 h, the population of hN2TM cells within any given culture well had heterogeneous morphological characteristics. The number of neurites per neuron, as well as total neurite length per neuron, varied from cell to cell. The ArrayScan VTI HCS reader allows the user to define a minimum number of neurons to be sampled in each well (sampling threshold), and therefore control the number of cells used to calculate well-level averages of neurite count and total neurite length per neuron. Retrospective analyses of hN2TM cells at a density of 7500 cells/well demonstrated that the across well coefficient of variation (C.V.) for these two endpoints (n = 6 wells) dropped from 20% to less than 10% when the sampling threshold was increased from 10 cells (sampling a single field) to 100 cells (sampling 6–7 fields). Increasing the sampling threshold greater than 100 neurons per well did not appreciably lower the across well C.V.s further (data not shown). For all studies shown here, a sampling threshold of 350 neurons per well was used. In control experiments using this sampling threshold, measurements of the average number of neurites per neuron and total neurite length per neuron were very reproducible between cultures, varying by less than 6% at 24 h. 3.3. Chemical effects on hN2TM neurite outgrowth DMSO at concentrations between 0.05 and 0.5% had no effect on ATP-content, neuron density, the average number of neurites per neuron or total neurite length per neuron in hN2TM cells following 22 h of exposure (Fig. 5A–C). At a concentration of 1%, DMSO significantly decreased total neurite length per neuron by 21.7% (control: 44.1 6.6 mm, treated: 27.8 7.5 mm, Fig. 5C) with no significant effects on ATP-content, neuron density or the average number of neurites per neuron (Fig. 5A and B). At a concentration of 5% DMSO, a significant decrease in ATP-content (51.7%) and neuron density (63.2%) was observed coupled with significant decreases (>97%) in the average number of neurites and total neurite length per neuron. In the present study, DMSO was used to dissolve Bis1, U0126 and brefeldin A prior to preparation of dosing solutions. The final concentration of DMSO in tissue culture wells treated with these compounds did not exceed 0.1%. At this concentration of DMSO, no significant effects on ATP-content, neuron density, or neurite outgrowth measurements were observed. A set of five compounds with well defined molecular mechanisms of action was identified from the literature as having effects on neurite outgrowth in primary rodent neural cultures. Descriptions of these studies are listed in Table 1. For each compound a five point concentration-response curve was examined. Concentration ranges were based on exposure levels used in previous research as described in the literature (Table 1). Following a 22 h exposure, the protein kinase C (PKC) inhibitor Bis1 had no significant effects on ATP-content or neuron density at any of the concentrations examined (Fig. 6A). The threshold concentration of Bis1 for decreasing ATP-content or neuron density could not be determined from these data. In contrast, concentration-dependent decreases in neurite outgrowth was observed following exposure to Bis1 (Fig. 6B). At 10 mM a significant (22.3%) decrease in the average number of neurites per neuron (control: 1.14 0.22, treated: 0.97 0.13) as well as a significant (26%) decrease in total neurite length per neuron (control: 38.8 11.8 mm, treated: 31.9 4.5 mm) was observed. Significant decreases in neurite outgrowth were not observed at concentrations of Bis1 below 3 mM. These data demonstrate a specific inhibition of neurite outgrowth by 10 mM Bis1, a concentration that does not affect indicators of cell health. In contrast to Bis1, significant decreases in neurite outgrowth were accompanied by concurrent decreases in ATP-content and neuron density following exposure to U0126, an inhibitor of mitogen-activated protein kinase/extracellular-regulated kinase (MEK) signaling (Fig. 6C and D). There were no concentrations of U0126 that affected neurite outgrowth without a concurrent effect on either ATP-content or neuron density. Between 3 and 30 mM of U0126, ATP-content was significantly decreased by approximately 20% from control values. A significant decrease in neuron density J.A. Harrill et al. / NeuroToxicology 31 (2010) 277–290 283 Fig. 3. Antigenic characterization of hN2TM cells. Cells grown in 96-well titer plates at 7500 cells/well (2.5 104 cells/cm2) for 24 h and immunocytochemically labeled for (A) bIII-tubulin, (B) nestin, (C) phosphorylated neurofilaments (pNFs) or (D) MAP2. Panels E–H are modulation contrast images corresponding to fluorescently imaged fields to the immediate left each panel. All images were taken at 20 magnification on a Leica DMI6000 microscope. All cell bodies and neurites were positive for both bIII-tubulin and nestin at 24 h. Arrows in panels (C) and (G) correspond to neurites that are not positive for pNFs, a marker of neuronal axons. Likewise, arrows in panels (D) and (H) correspond to neurites that are not positive for MAP2, a marker of neuronal dendrites. Scale bars = 100 mm. 284 J.A. Harrill et al. / NeuroToxicology 31 (2010) 277–290 Fig. 4. Effects of time and plating density on neurite outgrowth in hN2TM cells. (A–C) Cells were grown for either 2, 6, 24 or 48 h at a density of 2500 to 10,000 cells/well (8.33 103 and 3.33 104 cells/cm2) and measured. (A) Average number of neurites per neuron. (B) Normalized histogram of the average number of neurites per neuron. (C) Total neurite length per neuron. Values for each measurement significantly increased over time between 2 and 48 h (p < 0.05, one-way ANOVA). (D–F) Cells were grown for 24 h at densities of 2500, 5000, 7500 or 10,000 cells/cm2 and measured. (D) Average number of neurites per neuron. (E) Normalized histogram of the average number of neurites per neuron. (F) Total neurite length per neuron. Values for each measurement were not significantly different across this range of plating densities. n = 10–12 wells from two independent cultures for both time course and plating density experiments. (29.2%) was observed only at 30 mM, 10-fold higher than the minimum concentration which decreased ATP-content (Fig. 6C). Decreases in neurite outgrowth were first observed at 10 mM U0126 and continued up to 30 mM, the highest concentration tested (Fig. 6D). At 30 mM, significant decreases of 45% (control: 1.14 0.22, treated: 0.72 0.33) and 53.2% (control: 38.8 11.8 mm, treated: 22.6 12 mm) were observed for the average number of neurites per neuron and total neurite length per neuron, respectively. Concentrations of U0126 between 0.3 and 1 mM had no significant effects on ATP-content, neuron density or neurite outgrowth measurements. Exposure to millimolar concentrations of lithium chloride (LiCl) also affected hN2TM ATP-content, neuron density and neurite outgrowth (Fig. 6E and F). At 10 mM and 30 mM LiCl, ATP-content was significantly decreased by 27.1 and 45.6%, respectively, compared to untreated controls. Neuron density was also significantly decreased by 20.3 and 56.3% at these concentrations, respectively. At 10 mM, significant decreases of 49.1% (control: 1.13 0.23, treated: 0.59 0.15) and 62% (control: 38.6 12.1 mm, treated: 14.8 4.3 mm) were observed for the average number of neurites per neuron and total neurite length per neuron, respectively. At 30 mM, both the average number of neurites per neuron and total neurite length per neuron were significantly decreased >90% from vehicle treated control values. Similar to the patterns observed with U0126, there were no concentrations of LiCl that affected neurite outgrowth without concurrent effects ATP-content or neuron density. Similarly, the fungal antibiotic brefeldin A significantly decreased neurite outgrowth but only at concentrations where concurrent significant decreases in ATP-content and neuron density were observed (Fig. 6G and H). This compound disrupts membrane transport of proteins from intracellular organelles (i.e. endoplasmic reticulum and Golgi) to the cell membrane. The concentration-response for brefeldin A on both neurite outgrowth endpoints was steep, with no effects observed between 0.01 and 0.03 mM and significant decreases (>85%) at concentrations ranging from 0.1 to 1 mM. In contrast, significant decreases in ATP-content and neuron density were observed at the same concentrations (0.1–1 mM) but only to 50% of control levels. The effects of the broad-spectrum phosphatase inhibitor Na3VO4 were unique among the compounds examined in that significant concentration-dependent decreases in all endpoints were observed with marked differences in the lowest effective concentrations that inhibited neurite outgrowth, neuron density and ATP-content, respectively (Fig. 6I and J). Significant decreases in neurite outgrowth measurements were first observed at 3 mM Na3VO4, whereas significant decreases in hN2TM neuron density and ATP-content were first observed at 10 and 30 mM, respectively. Following exposure to 10 mM Na3VO4, significant decreases of 36.8% (control: 1.23 0.14, treated: 0.77 0.08) and 47.8% (control: 42.7 8.2 mm, treated: 21.9 3.2 mm) were observed for the average number of neurites and total neurite length per neuron, respectively (Fig. 6J). Neuron density significantly decreased by 10 mM Na3VO4 (22.1%) and ATP-content was not affected at this concentration (Fig. 6I). Neurite outgrowth measurements were significantly decreased by >94% by 30 and 100 mM Na3VO4 with concurrent, significant decreases in ATP-content (47.7 and 73%, respectively) and neuron density (65.0 and 76.8%, respectively). Collectively, these data demonstrate that neurite outgrowth measurements were more sensitive than indicators of cell health for detecting concentration-dependent effects of Na3VO4 on hN2TM cells. 4. Discussion The present study characterized the molecular phenotype of hESC-derived hN2TM cells grown in dissociated culture and quantified neurite outgrowth in these cells. At 24 h after initial plating, the cells expressed protein markers characteristic of maturing neurons. This is consistent with development along a neural lineage. Automated high-content image analysis (HCA) demonstrated that neurite outgrowth progressed rapidly once J.A. Harrill et al. / NeuroToxicology 31 (2010) 277–290 Fig. 5. Effects of DMSO on hN2TM neurite outgrowth. Cells were grown at a density of 7500 cells/well and exposed to either 0.05, 0.1, 0.5, 1 or 5% DMSO at 2 h after plating. Measurements were made at 24 h. (A) ATP-content (white bars) and neuron density (i.e. the average number of neurons per field; gray bars) were measured as indicators of cell health. (B) Average number of neurites per neuron. (C) Total neurite length per neuron. All data are presented as % change from untreated control wells standard deviation (S.D.). ATP-content data is from three separate experiments using independent cultures (control group, n = 27 wells total; treatment groups, n = 11–17 wells total). Neuron density and neurite outgrowth data is from two separate experiments using independent cultures (control group, n = 24 wells total; treatment groups, n = 10–16 wells total). A significant effect of concentration was observed for each endpoint (p < 0.05, one-way ANOVA). *Concentration is significantly different from control values (p < 0.01, Dunnett’s post hoc test). cells were plated on a poly-L-lysine/laminin substrate. Within 24 h of initiating the cultures, a large proportion of cells had developed one to three neurites. Finally, exposure to a panel of chemicals known to affect the growth of neurites in rodent primary neural cultures produced concentration-dependent decreases in neurite outgrowth in these human neural cells. Collectively, these data indicate that hN2TM cells have morphological and phenotypic characteristics of maturing neurons and are amenable to measurement of neurite outgrowth using medium-throughput HCA methods. Immunocytochemical staining demonstrated that at 24 h after initial plating, hN2TM cells expressed markers indicative of neuroepithelial lineage. Prior to initiation of cultures by the end 285 user, the cells have been subjected to culture conditions designed to promote neuronal differentiation. Accordingly, during HCA analysis, the average number of cell nuclei per field did not increase over time (data not shown) indicating that these cells were in a non-proliferative state, a feature of differentiated ESCs. In addition, the cells expressed nestin (Fig. 2), an intermediate filament protein expressed in human neuroepithelium in vivo and hESC-derived neuroepithelial cells in culture (Tohyama et al., 1992; Gilyarov, 2008). In the developing embryonic nervous system in vivo, nestin expression decreases as cells become postmitotic (Dahlstrand et al., 1995). Similarly, nestin expression also decreases in vitro as ESCs mature into a non-proliferative state (Rolletschek et al., 2001). However, in vitro, some cells may remain positive for nestin protein for up to several weeks following stimuli that promote neuronal differentiation (Shin et al., 2006; Rolletschek et al., 2001; Nat et al., 2007). Therefore, positive nestin expression suggested that hN2TM cells are of a neuroepithelial lineage, but does not indicate which stage of neural differentiation these cells were in shortly after initiation of cultures from frozen stocks. In addition to nestin, hN2TM cells were also positive for bIIItubulin, MAP2 and phosphorylated neurofilaments (Fig. 2), all of which are expressed in mature neurons in vitro (Fletcher and Banker, 1989; Caceres et al., 1986; DeFuria and Shea, 2007). Expression of bIII-tubulin and MAP2 increases in ESCs upon differentiation along a neural lineage and have been found to be coexpressed with nestin following a differentiating stimulus (Nat et al., 2007; Baharvand et al., 2007). In addition, the patterns of MAP2 and phosphorylated neurofilament immunolabeling (Fig. 2E–H) indicated that hN2TM cells grown for 24 h displayed early features of neuronal polarity, a fundamental functional property of mature neurons. Neuronal polarization facilitates the unidirectional flow of electrical activity from synaptic contacts on the dendritic arbor and cell body to neurotransmitter release sites in the axon (Arimura and Kaibuchi, 2007). Expression of MAP2 protein is restricted to the dendritic compartment while the presence of a number of phosphorylated neurofilaments (detected by the SMI-312 antibody) is specific to axons (Ulfig et al., 1998; Kosik and Finch, 1987). Fig. 3 demonstrates that some hN2TM cells grown for 24 h had some neurites which were MAP2-positive and others which were MAP2-negative. Likewise, some neurites were positive for pNFs while others were negative. These staining patterns demonstrated cytoplasmic compartmentalization and indicate that these cells may have been developing toward a polarized neuronal phenotype. Dual-immunolabeling experiments using these cells are needed to clarify if neurites selectively labeled with MAP2 and pNFs can develop from the same cell. Overall, it is clear from the present data that hN2TM cells plated from frozen stocks had a molecular phenotype consistent with developing neurons. Staining patterns indicate that these cells are in a developmental period between the onset of mitotic quiescence and terminal differentiation into a mature, polarized neuron at 24 h after plating. Dissociated cultures of hN2TM cells immunostained for bIIItubulin were amenable to measurement of neurite outgrowth using automated HCA. In order to accurately perform neurite outgrowth measurements using HCA, cells must be grown at a low enough density so that the morphological features of individual cells can be easily resolved (Dragunow, 2008). In the present experiment, increasing the plating density from 2500 to 10,000 cells/well had no significant effect on measurements of neurite outgrowth (Fig. 4). Within this range the morphological features of individual cells could be easily resolved and the variability in population mean measurements was low, both from well-to-well and experiment-to-experiment. Above 10,000 cells/ well, the complexity of the image prevented accurate assignment 286 J.A. Harrill et al. / NeuroToxicology 31 (2010) 277–290 Fig. 6. Chemical effects on neurite outgrowth in hN2TM cells. Cells were grown at a density of 7500 cells/cm2 and exposed to varying concentrations of Bis1 (A and B), U0126 (C and D), LiCl (E and F), brefeldin A (G and H) or Na3VO4 (I and J) 2 h after plating. Measurements were made at 24 h after plating. ATP-content (open circles) and neuron density (i.e. average number of neurons per field, open squares) are presented in the left-hand column (A, C, E, G, and I). Average number of neurites per neuron (open circles) and total neurite length per neuron (open squares) are presented in the right-hand column (B, D, F, H, and J). Note, the x-axis for LiCl (panels G and H) is in mM as opposed to mM. All data are presented as % change from untreated control (LiCl, Na3VO4) or controls containing 0.1% DMSO (Bis1, U0126, brefeldin A) standard deviation (S.D.). For Bis1, U0126, LiCl and Na3VO4 ATP-content, neuron density and neurite outgrowth data is from 2 separate experiments using independent cultures (control group, n = 9–15 wells total; treatment groups, n = 6–9 wells total). For brefeldin A, data is from 3 separate experiments using independent cultures (control group, n = 24–27 wells; treatment groups, n = 5–11 wells). A significant main effect of concentration was found for each endpoint for all compounds tested, except ATP-content and neuron density with Bis1 (p < 0.05, one-way ANOVA). Symbols above, below or immediately beside data points denotes that the response at a given concentration is significantly different from control values for ATP-content (*), neuron density (**), average number of neurites per neuron (#), total neurite length per neuron (§) using Dunnett’s post hoc test (p < 0.01). J.A. Harrill et al. / NeuroToxicology 31 (2010) 277–290 of neurites to associated cell bodies (data not shown) and precluded automated measurements. These data demonstrated that during the early growth phase, the density of the cultures had no influence on the rate of neurite outgrowth. Similarly, Radio et al. (2010) demonstrated that neurite outgrowth in an NGF-stimulated PC12 cell clone (Neuroscreen-1, NS-1) was not affected by changes in plating density whereas neurite outgrowth in a primary mixed neural culture was dramatically affected. In the latter type of culture model, neurite outgrowth is influenced by cell–cell contacts as well as soluble factors and extracellular matrix proteins secreted by supportive glial cell populations (Hatten et al., 1988; Ibata et al., 1991). The cultures in the present study appeared to be of a homogeneous neuronal phenotype. The morphological changes observed in hN2TM cells over time were also consistent with these cells developing toward a neuronal phenotype. Neurite outgrowth progressed rapidly during the first 48 h of growth (Fig. 4). Greater than 60% of the cells had developed between one and three neurites by 24 h after plating. The rate and total amount of neurite outgrowth was comparable to that of clonal neural cell lines and primary neural cultures cited in the literature (Radio et al., 2010; Bearer et al., 1999; Hong et al., 2003; Das et al., 2004; Vutskits et al., 2006; George et al., 2009). From these data, it is clear that morphological development of hN2TM cells progressed rapidly and was similar to that observed in previously characterized neural culture models. Therefore, these types of human stem cell-derived neural cells provide an alternative to rodent primary cultures or immortalized clonal cell lines for investigating the mechanisms of neurite outgrowth. Neurite outgrowth in hN2TM cells was inhibited by a variety of chemicals previously shown to affect neurite outgrowth in primary neural culture models (Table 1). In developing neurons, neurite outgrowth is dependent upon a number of inter-connected cellular processes including membrane and protein trafficking, reorganization of the cytoskeleton and a variety of intracellular signaling cascades (Larsson, 2006; Yoshimura et al., 2006; Tang, 2001; Dent and Gertler, 2003; Ditlevsen et al., 2008). In the present study, a diverse set of test chemicals that disrupt these processes were examined in order to compare and contrast neurite outgrowth in hESC-derived neural cells with that of previously established models. Overall, the present data demonstrated that these hESCderived cells were sensitive to agents that disrupt neurite outgrowth in primary rodent neural cultures. In addition, these data demonstrated that the molecular mechanisms that mediate neurite outgrowth in neural clonal cell lines and rodent primary cultures may also play a role in the developmental biology of hESCderived neurons (Table 2). In the present study two measures were used as general indicators of cell health: ATP-content as a measure of metabolic status and neuron density (i.e. the average number of neurons per field) as a measure of cell viability. These data were coupled with measurements of neurite outgrowth to provide a context in which to evaluate of the overall health of the cultures when treatment- 287 related decreases in neurite outgrowth were observed. In the context of quantitative screening for neurotoxicants, measurements of cell health or cytotoxicity alone may not be sufficient to: (1) identify neurotoxic compounds or (2) provide accurate information on the potency for eliciting a neurotoxic response. For example, in the former case, ATP-content measurements were unable to positively identify trans-retinoic acid, a known developmental neurotoxicant (DNT), as having any detrimental effects on PC12 cells in the absence of neurite outgrowth data (Radio et al., 2008, 2010). In the latter case, the threshold for effects on ATPcontent for the known developmental neurotoxicant methylmercury (MeHg) was 10,000-fold greater than the minimum effective concentration needed to affect neurite outgrowth in PC12 cells (Radio et al., 2008, 2010). These data suggest that measurements of neurite outgrowth may be more sensitive than cell health assays, and thus have a greater capability for accurate identification of potential developmental neurotoxicants. In the present study, a majority of the chemicals tested (3 out of 5 as outlined above) produced decreases in neurite outgrowth measurements only at concentrations that also affected ATPcontent and/or neuron density. The relative sensitivity of these two endpoints varied across compounds. However, the patterns of concentration-dependent effects were consistent across endpoints for each compound tested. In instances where effects on neurite outgrowth were detected the relative magnitude of the effect was greater than that observed with ATP-content or neuron density measurements. This supports the hypothesis that measurements of neurite outgrowth are more sensitive than measures of cell health in the context of neurotoxicity screening. In addition, the effects observed following Na3VO4 and Bis1 treatment confirms that a neuronal cell-type specific toxicity response can be observed in the absence of acute changes in indicators of cell health in hESCderived cells. Concentration-response data suggested that the PKC and MEK/ ERK signaling pathways may mediate neurite outgrowth in hN2TM cells. The PKC inhibitor Bis1 decreased the average number of neurites and total neurite length per neuron with no observable effects on ATP-content or neuron density in the concentration range examined (Fig. 6A and B). U0126, an inhibitor of MEK/ERK signaling, also decreased both measures of neurite outgrowth, but only where concurrent decreases in ATP-content or neuron density were observed (Fig. 6C and D). Both the PKC and the MEK/ERK signaling pathways have been shown to mediate neurite outgrowth in both clonal and primary neural culture models following application of neurotrophic receptor ligands (i.e. nerve growth factor, NGF) and cell adhesion molecules (Kim et al., 1997; Gerecke et al., 2004; Schmid et al., 1999; Burry, 1998; Kolkova et al., 2000). Specifically, Bis1 and U0126 dramatically inhibited neurite outgrowth in NGF-stimulated PC-12 cells as well as primary cerebellar granule cells (CGCs) and carbachol-stimulated primary hippocampal neurons (Radio et al., 2010; Das et al., 2004; VanDeMark et al., 2009). Importantly, Radio et al. (2010) Table 2 Summary of concentration-response data. Chemical Brefeldin A Bis1 U0126 Na3VO4 LiCl Concentration range 0.01–1 mM 0.1–10 mM 0.3–30 mM 1–100 mM 300–30,000 mM Lowest effective concentrationa ATP content Neuron densityb Average # of neurites per neuron Total neurite length per neuron 0.1 n.d. 30 30 10,000 0.1 n.d. 3 10 10,000 0.1 10 10 3 10,000 0.1 10 10 3 10,000 a Values are the lowest effective concentration (in mM) for which a significant change from control was detected using a Dunnett’s many-to-one mean contrast test (p < 0.01). n.d. = no change detected. b Average number of neurons per field. 288 J.A. Harrill et al. / NeuroToxicology 31 (2010) 277–290 demonstrated that the characteristics of neurite outgrowth inhibition by these agents may vary from cell-type to cell-type. In PC12 (NS-1) cells Bis1 inhibited neurite outgrowth with no apparent decreases in ATP-content (i.e. cell health) indicating a specific effect on this developmental process. In contrast, neurite outgrowth inhibition by Bis1 in primary CGCs occurred only at concentrations where ATP-content was also decreased, indicating the inhibition was likely a secondary response to decreased cell health (Radio et al., 2010). These data demonstrate that parallel analysis of neurite outgrowth with some measure of cell health can be critical for interpretation of neurite outgrowth data from in vitro neurotoxicity studies. The present data support that inhibition of hN2TM neurite outgrowth by 10 mM Bis1 is not a secondary response to a decrease in cell health and also indicates that PKC signaling is likely involved in neurite outgrowth in hESCderived neurons. In contrast, it is unclear from the present data whether U0126 decreased neurite outgrowth in hN2TM cells as a specific result of MEK/ERK inhibition or whether the decreases in neurite outgrowth were the result of a generalized decrease in cell health in response to this compound. Collectively, the present data indicate that the PKC pathway likely plays a role in mediating neurite outgrowth in hESC-derived neurons and that the MEK/ERK pathway is active in the maintenance of cell health. Additional studies are required to define and refine the specific role of the PKC and MEK/ERK signaling pathways in neurite outgrowth of hESCderived neurons. In addition to kinase inhibitors, the protein tyrosine phosphatase inhibitor Na3VO4 also decreased neurite outgrowth in hN2TM cells. Na3VO4 acts as a broad-spectrum tyrosine phosphatase inhibitor and decreases axonal outgrowth in hippocampal neurons and neurite outgrowth in NGF-stimulated PC12 cells (Wu and Bradshaw, 1993; Mandell and Banker, 1996). The present data are consistent with these previous findings. In addition, decreases in neurite outgrowth were observed at concentrations of Na3VO4 that did not produce decreases in neuron density or ATP-content (Fig. 6I and J). These data demonstrate that at 3 mM the effects of Na3VO4 were specific to the process of neurite outgrowth. A similar outcome was observed with Bis1 at a concentration of 10 mM. As the concentration of Na3VO4 increased the effects on neurite outgrowth were accompanied by decreases in neuron density at 10 mM and ATP-content at 30 mM. These data reflect a progression in the severity Na3VO4 toxicity from a specific effect on a developmental process to a more generalized effect on cell health. These data demonstrated that coupling measurements of neurite outgrowth with indicators of cell health permits detection of chemical effects on neuronal morphology that are not the result of generalized decreases in cell health. These data also imply that for some compounds neurite outgrowth may be a more sensitive endpoint for detecting developmental neurotoxicity potential as compared to cell counts, measures of ATP-content or other indicators of cell health. From these data, Na3VO4 or Bis1 may serve as an endpoint specific control for neurite outgrowth in hESC-derived neural cells for toxicity screening applications. Brefeldin A, a fungal antibiotic that disrupts membrane trafficking, produced a decrease in neurite outgrowth in hN2TM cells (Fig. 6G and H). Decreases in neurite outgrowth were accompanied by concurrent 30–40% decreases in ATP-content and neuron density. Brefeldin A acts by inhibiting a guanine nucleotide exchange factor that regulates the activity of ADP-ribosylation factor (ARF). ARF controls membrane trafficking from the Golgi to the plasma membrane (Donaldson et al., 1992). Treatment of neurons with brefeldin A terminates membrane trafficking and eventually results in dissolution of the Golgi membrane network. This results in a reduction in the rate of neurite extension and, in some cases, neurite retraction (Jareb and Banker, 1997; PragerKhoutorsky and Spira, 2009). The present data indicate that membrane trafficking from the Golgi is necessary for promoting neurite outgrowth in hN2TM cells and maintaining cell health, which is consistent with observations from primary rodent neural cell cultures. Neurite outgrowth in hN2TM cells was also inhibited by 10– 30 mM concentrations of lithium chloride (LiCl). Neurite outgrowth was inhibited >49% from control values and was accompanied by concurrent decreases in ATP-content and neuron density (Fig. 6E and F). This is consistent with data from Takahashi et al. (1999) which also demonstrates concurrent decreases in neurite outgrowth and cell viability in primary rat hippocampal cultures following LiCl exposure. In contrast, there are a number of conflicting reports in the literature regarding the effect LiCl on neurite outgrowth. Some studies in primary neural cultures and clonal cell lines demonstrate an inhibition of neurite outgrowth with mM concentrations of LiCl (Takahashi et al., 1999; Koike et al., 2006; Hollander and Bennett, 1991; Tamura and Ohkuma, 1991) while others demonstrate enhancement (Orme et al., 2003; Sayas et al., 2002) or even a biphasic effect (Munoz-Montano et al., 1999). The source of the disparate findings between studies is unknown, but could be due to differences in cell-types or culture conditions across studies. In the context of neurite outgrowth, the putative molecular mechanism-of-action of LiCl is inhibition of kinases that regulate cytoskeletal protein dynamics such as glycogen-synthase kinase 3b (GSK3b) (Yoshimura et al., 2006) and nemo-like kinase (NLK) (Ishitani et al., 2009). The expression patterns, activity levels and sub-cellular distribution of GSK3b and NLK have not been characterized in hESC-derived neural cells. However, the inhibition of neurite outgrowth by LiCl suggests a putative role for these kinases in mediating this process. Alternatively, the decrease in neurite outgrowth observed here could be due a decrease in general cell health and off-target effects caused by mM concentrations of LiCl. Characterizing the expression of GSK3b and NLK and examining neurite outgrowth in response to protein knockdown or more specific pharmacological inhibitors would aid in defining the role of these proteins in development of hESC-derived neural cells. Interestingly, decreases in hN2TM neurite outgrowth were observed with both selective (Bis1, U0126) and non-selective (LiCl) kinase inhibitors as wells as a non-selective phosphatase inhibitor (Na3VO4). In general, protein phosphorylation acts a molecular switch controlling the activity of a large variety intracellular signaling pathways and processes. In the case of neurite outgrowth, phosphorylation of effector proteins can act to either promote or inhibit neurite outgrowth depending upon the function of the protein. For example, phosphorylation of microtubule-associated protein 1B (MAP1B) downstream of GSK3b and NLK and phosphorylation of paxillin downstream of NLK promotes neurite outgrowth in NGF-stimulated PC12 cells (Ishitani et al., 2009). In contrast, phosphorylation of collapsin response mediator protein 2 (CRMP2) downstream of GSK3b inhibits neurite development in hippocampal neurons and correlates with a decrease in neurite outgrowth in NGFstimulated PC12 cells (Yoshimura et al., 2005; Patrakitkomjorn et al., 2008). Neurite outgrowth in hESC-derived neural cells may be mediated by a similar phenomenon; i.e. the balance of phosphorylation-dependent activity and inactivity of stimulatory and inhibitory effector proteins. The present data support that chemicals that interfere with phosphorylation and dephosphorylation of proteins may result in similar inhibitory effects on neurite outgrowth in hESC-derived neural cells, possibly by disrupting this type of balanced intracellular signaling network. More detailed biochemical studies are required to identify the phosphorylation-dependent effectors of neurite outgrowth and define the molecular mechanisms whereby chemicals can inhibit this developmental process in hESC-derived neural cells. J.A. Harrill et al. / NeuroToxicology 31 (2010) 277–290 5. Conclusions In summary, this work demonstrates that the molecular phenotype and processes controlling neurite outgrowth in hN2TM cells are similar to previously characterized neuronal cell models in vitro. In contrast to rodent primary neural cultures, hN2TM cells provide a model in which to investigate the processes of neurite outgrowth in the context of human biology. In addition, the cells are non-transformed and provide an alternative to the use of tumor-derived or transfected clonal cell lines. These cells have several qualities that make them amenable for use in high- to medium-throughput developmental neurotoxicity screening, including: cellular homogeneity, a rapid rate of neurite outgrowth, low inter-experiment variability in automated morphological measurements and the ability to be cultured at low densities. These data also demonstrate Na3VO4 or Bis1 could serve as an endpoint specific positive controls for inhibition of neurite outgrowth. Additional studies are needed to characterize the molecular pathways controlling neurite outgrowth in hESCderived neural cells and whether these cells continue to mature and form functional networks in culture. In the context of neurotoxicity screening, future efforts will include evaluation of a larger training set of inert and known DNT compounds in order to assess the sensitivity of the hN2TM culture model as a DNT screening tool. Conflict of interest statement The data included in this study was generated at the U.S. Environmental Protection Agency, National Health and Environmental Effects Research Laboratories. hN2TM cells and growth media were provided through Material Transfer Agreement #46608 between the U.S. EPA and ArunA Biomedical, Inc. There is a potential conflict of interest in that one author (Steven L. Stice) currently serves as the Chief Scientific Officer of ArunA Biomedical, Inc. and another author (Dave Machacek) was employed by ArunA Biomedical, Inc. during the study. Acknowledgements The authors wish to thank Mr. Brian Robinette for his technical assistance and thank Drs. Will Boyes and Linda Kaltenbach for their comments and suggestions on an earlier version of this manuscript. References Allen DD, Caviedes R, Cardenas AM, Shimahara T, Segura-Aguilar J, Caviedes PA. Cell lines as in vitro models for drug screening and toxicity studies. Drug Dev Ind Pharm 2005;31(8):757–68. Arimura N, Kaibuchi K. Neuronal polarity: from extracellular signals to intracellular mechanisms. Nat Rev Neurosci 2007;8(3):194–205. Audesirk T, Audesirk G, Ferguson C, Shugarts D. Effects of inorganic lead on the differentiation and growth of cultured hippocampal and neuroblastoma cells. Neurotoxicology 1991;12(3):529–38. Baharvand H, Mehrjardi NZ, Hatami M, Kiani S, Rao M, Haghighi MM. Neural differentiation from human embryonic stem cells in a defined adherent culture condition. Int J Dev Biol 2007;51(5):371–8. Bearer CF, Swick AR, O’Riordan MA, Cheng G. Ethanol inhibits L1-mediated neurite outgrowth in postnatal rat cerebellar granule cells. J Biol Chem 1999;274(19): 13264–70. Breier JM, Gassmann K, Kayser R, Stegeman H, De Groot D, Fritsche E, et al. Neural progenitor cells as models for high-throughput screens of developmental neurotoxicity: state of the science. Neurotoxicol Teratol 2009. Burry RW. PKC activators (phorbol ester or bryostatin) stimulate outgrowth of NGFdependent neurites in a subline of PC12 cells. J Neurosci Res 1998;53(2):214–22. Caceres A, Banker GA, Binder L. Immunocytochemical localization of tubulin and microtubule-associated protein 2 during the development of hippocampal neurons in culture. J Neurosci 1986;6(3):714–22. Cedrola S, Guzzi G, Ferrari D, Gritti A, Vescovi AL, Pendergrass JC, et al. Inorganic mercury changes the fate of murine CNS stem cells. FASEB J 2003;17(8):869–71. 289 Coecke S, Goldberg AM, Allen S, Buzanska L, Calamandrei G, Crofton K, et al. Workgroup report: incorporating in vitro alternative methods for developmental neurotoxicity into international hazard and risk assessment strategies. Environ Health Perspect 2007;115(6):924–31. Costa LG, Aschner M, Vitalone A, Syversen T, Soldin OP. Developmental neuropathology of environmental agents. Annu Rev Pharmacol Toxicol 2004;44:87–110. Dahlstrand J, Lardelli M, Lendahl U. Nestin mRNA expression correlates with the central nervous system progenitor cell state in many, but not all, regions of developing central nervous system. Brain Res Dev Brain Res 1995;84(1):109–29. Das KP, Freudenrich TM, Mundy WR. Assessment of PC12 cell differentiation and neurite growth: a comparison of morphological and neurochemical measures. Neurotoxicol Teratol 2004;26(3):397–406. DeFuria J, Shea TB. Arsenic inhibits neurofilament transport and induces perikaryal accumulation of phosphorylated neurofilaments: roles of JNK and GSK-3beta. Brain Res 2007;1181:74–82. Dent EW, Gertler FB. Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron 2003;40(2):209–27. Ditlevsen DK, Povlsen GK, Berezin V, Bock E. NCAM-induced intracellular signaling revisited. J Neurosci Res 2008;86(4):727–43. Donaldson JG, Finazzi D, Klausner RD. Brefeldin A inhibits Golgi membrane-catalysed exchange of guanine nucleotide onto ARF protein. Nature 1992;360(6402): 350–2. Dragunow M. High-content analysis in neuroscience. Nat Rev Neurosci 2008;9(10): 779–88. Fletcher TL, Banker GA. The establishment of polarity by hippocampal neurons: the relationship between the stage of a cell’s development in situ and its subsequent development in culture. Dev Biol 1989;136(2):446–54. George J, Dravid SM, Prakash A, Xie J, Peterson J, Jabba SV, et al. Sodium channel activation augments NMDA receptor function and promotes neurite outgrowth in immature cerebrocortical neurons. J Neurosci 2009;29(10):3288–301. Gerecke KM, Wyss JM, Carroll SL. Neuregulin-1beta induces neurite extension and arborization in cultured hippocampal neurons. Mol Cell Neurosci 2004;27(4): 379–93. Gilyarov AV. Nestin in central nervous system cells. Neurosci Behav Physiol 2008;38(2):165–9. Grandjean P, Landrigan PJ. Developmental neurotoxicity of industrial chemicals. Lancet 2006;368(9553):2167–78. Harry GJ, Tiffany-Castiglioni E. Evaluation of neurotoxic potential by use of in vitro systems. Expert Opin Drug Metab Toxicol 2005;1(4):701–13. Hatten ME, Lynch M, Rydel RE, Sanchez J, Joseph-Silverstein J, Moscatelli D, et al. In vitro neurite extension by granule neurons is dependent upon astroglial-derived fibroblast growth factor. Dev Biol 1988;125(2):280–9. Higgins D, Banker G. Primary dissociated cell cultures. In: Stevens CF, Banker G, Goslin K, editors. Culturing nerve cells 2nd ed. Cambridge, MA: MIT Press; 1998. Hollander BA, Bennett GS. Lithium chloride alters cytoskeletal organization in growing, but not mature, cultured chick sensory neurons. J Neurosci Res 1991;28(3): 332–42. Hong MS, Hong SJ, Barhoumi R, Burghardt RC, Donnelly KC, Wild JR, et al. Neurotoxicity induced in differentiated SK-N-SH-SY5Y human neuroblastoma cells by organophosphorus compounds. Toxicol Appl Pharmacol 2003;186(2):110–8. Howard AS, Bucelli R, Jett DA, Bruun D, Yang D, Lein PJ. Chlorpyrifos exerts opposing effects on axonal and dendritic growth in primary neuronal cultures. Toxicol Appl Pharmacol 2005;207(2):112–24. Ibata Y, Toya S, Kohsaka S. Effects of astroglia on the development of cultured neurons from embryonic rat cerebral cortex. Comp Biochem Physiol C 1991;98(1):139–46. Ishitani T, Ishitani S, Matsumoto K, Itoh M. Nemo-like kinase is involved in NGFinduced neurite outgrowth via phosphorylating MAP1B and paxillin. J Neurochem 2009;111(5):1104–18. Jareb M, Banker G. Inhibition of axonal growth by brefeldin A in hippocampal neurons in culture. J Neurosci 1997;17(23):8955–63. Jin X, Hu H, Mathers PH, Agmon A. Brain-derived neurotrophic factor mediates activitydependent dendritic growth in nonpyramidal neocortical interneurons in developing organotypic cultures. J Neurosci 2003;23(13):5662–73. Khaibullina AA, Rosenstein JM, Krum JM. Vascular endothelial growth factor promotes neurite maturation in primary CNS neuronal cultures. Brain Res Dev Brain Res 2004;148(1):59–68. Kim B, Leventhal PS, Saltiel AR, Feldman AR. Insulin-like growth factor-I-mediated neurite outgrowth in vitro requires mitogen-activated protein kinase activation. J Biol Chem 1997;272(34):21268–73. Koike T, Uno S, Ishizawa M, Takahashi H, Ikeda K, Yokota S, et al. The heat shock protein inhibitor KNK437 induces neurite outgrowth in PC12 cells. Neurosci Lett 2006;410(3):212–7. Kolkova K, Novitskaya V, Pedersen N, Berezin V, Bock E. Neural cell adhesion molecule-stimulated neurite outgrowth depends on activation of protein kinase C and the Ras-mitogen-activated protein kinase pathway. J Neurosci 2000;20(6): 2238–46. Kosik KS, Finch EA. MAP2 and tau segregate into dendritic and axonal domains after the elaboration of morphologically distinct neurites: an immunocytochemical study of cultured rat cerebrum. J Neurosci 1987;7(10):3142–53. Larsson C. Protein kinase C and the regulation of the actin cytoskeleton. Cell Signal 2006;18(3):276–84. Lein P, Gallagher PJ, Amodeo J, Howie H, Roth JA. Manganese induces neurite outgrowth in PC12 cells via upregulation of alpha(v) integrins. Brain Res 2000;885(2):220–30. Lein P, Silbergeld E, Locke P, Goldberg AM. In vitro and other alternative approaches to developmental neurotoxicity testing (DNT). Environ Toxicol Pharmacol 2005;19: 735–44. 290 J.A. Harrill et al. / NeuroToxicology 31 (2010) 277–290 LePage KT, Dickey RW, Gerwick WH, Jester EL, Murray TF. On the use of neuro-2a neuroblastoma cells versus intact neurons in primary culture for neurotoxicity studies. Crit Rev Neurobiol 2005;17(1):27–50. Mandell JW, Banker GA. Microtubule-associated proteins, phosphorylation gradients, and the establishment of neuronal polarity. Perspect Dev Neurobiol 1996;4(2–3): 125–35. McNeish J. Embryonic stem cells in drug discovery. Nat Rev Drug Discov 2004;3(1):70– 80. Moors M, Rockel TD, Abel J, Cline JE, Gassmann K, Schreiber T, et al. Human neurospheres as three-dimensional cellular systems for developmental neurotoxicity testing. Environ Health Perspect 2009;117(7):1131–8. Munoz-Montano JR, Lim F, Moreno FJ, Avila J, Diaz-Nido J. Glycogen synthase kinase-3 modulates neurite outgrowth in cultured neurons: possible implications for neurite pathology in Alzheimer’s disease. J Alzheimers Dis 1999;1(6):361–78. Nat R, Nilbratt M, Narkilahti S, Winblad B, Hovatta O, Nordberg A. Neurogenic neuroepithelial and radial glial cells generated from six human embryonic stem cell lines in serum-free suspension and adherent cultures. Glia 2007;55(4): 385–99. National Research Council, Board on Environmental Studies and Toxicology, Institute for Laboratory Animal Research, Institute for Laboratory Animal Research. Toxicity testing in the 21st century. A vision and a strategy. Washington, D.C.: National Academies Press; 2007. Orme MH, Giannini AL, Vivanco MD, Kypta RM. Glycogen synthase kinase-3 and Axin function in a beta-catenin-independent pathway that regulates neurite outgrowth in neuroblastoma cells. Mol Cell Neurosci 2003;24(3):673–86. Patrakitkomjorn S, Kobayashi D, Morikawa T, Wilson MM, Tsubota N, Irie A, et al. Neurofibromatosis type 1 (NF1) tumor suppressor, neurofibromin, regulates the neuronal differentiation of PC12 cells via its associating protein, CRMP-2. J Biol Chem 2008;283(14):9399–413. Prager-Khoutorsky M, Spira ME. Neurite retraction and regrowth regulated by membrane retrieval, membrane supply, and actin dynamics. Brain Res 2009;1251:65–79. Radio NM, Mundy WR. Developmental neurotoxicity testing in vitro: models for assessing chemical effects on neurite outgrowth. Neurotoxicology 2008;29(3):361–76. Radio NM, Breier JM, Shafer TJ, Mundy WR. Assessment of chemical effects on neurite outgrowth in PC12 cells using high content screening. Toxicol Sci 2008;105(1): 106–18. Radio NM, Freudenrich TM, Robinette BL, Crofton KM, Mundy WR. Comparison of PC12 and cerebellar granule cell cultures for evaluating neurite outgrowth using high content analysis. Neurotoxicol Teratol 2010;32(1):25–35. Redmond L, Kashani AH, Ghosh A. Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron 2002;34(6):999–1010. Reubinoff BE, Itsykson P, Turetsky T, Pera MF, Reinhartz E, Itzik A, et al. Neural progenitors from human embryonic stem cells. Nat Biotechnol 2001;19(12): 1134–40. Rice D, Barone S Jr. Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect 2000;108(Suppl. 3):511–33. Rolletschek A, Chang H, Guan K, Czyz J, Meyer M, Wobus AM. Differentiation of embryonic stem cell-derived dopaminergic neurons is enhanced by survivalpromoting factors. Mech Dev 2001;105(1–2):93–104. Sanes DH, Reh TA, Harriss WA. Axon growth and guidance. In: Development of the nervous system. London, UK: Elsevier Academic Press; 2006 p. 111–138. Sayas CL, Avila J, Wandosell F. Glycogen synthase kinase-3 is activated in neuronal cells by Galpha12 and Galpha13 by Rho-independent and Rho-dependent mechanisms. J Neurosci 2002;22(16):6863–75. Schmid RS, Graff RD, Schaller MD, Chen S, Schachner M, Hemperly JJ, et al. NCAM stimulates the Ras-MAPK pathway and CREB phosphorylation in neuronal cells. J Neurobiol 1999;38(4):542–58. Shin S, Dalton S, Stice SL. Human motor neuron differentiation from human embryonic stem cells. Stem Cells Dev 2005;14(3):266–9. Shin S, Mitalipova M, Noggle S, Tibbitts D, Venable A, Rao R, et al. Long-term proliferation of human embryonic stem cell-derived neuroepithelial cells using defined adherent culture conditions. Stem Cells 2006;24(1):125–38. Takahashi M, Yasutake K, Tomizawa K. Lithium inhibits neurite growth and tau protein kinase I/glycogen synthase kinase-3beta-dependent phosphorylation of juvenile tau in cultured hippocampal neurons. J Neurochem 1999;73(5):2073–83. Tamura H, Ohkuma S. Induction of neurite outgrowth of PC12 cells by an inhibitor of vacuolar H(+)-ATPase, bafilomycin A1. FEBS Lett 1991;294(1–2):51–5. Tang BL. Protein trafficking mechanisms associated with neurite outgrowth and polarized sorting in neurons. J Neurochem 2001;79(5):923–30. Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science 1998;282(5391):1145–7. Tohyama T, Lee VM, Rorke LB, Marvin M, McKay RD, Trojanowski JQ. Nestin expression in embryonic human neuroepithelium and in human neuroepithelial tumor cells. Lab Invest 1992;66(3):303–13. Ulfig N, Nickel J, Bohl J. Monoclonal antibodies SMI 311 and SMI 312 as tools to investigate the maturation of nerve cells and axonal patterns in human fetal brain. Cell Tissue Res 1998;291(3):433–43. VanDeMark KL, Guizzetti M, Giordano G, Costa LG. The activation of M1 muscarinic receptor signaling induces neuronal differentiation in pyramidal hippocampal neurons. J Pharmacol Exp Ther 2009;329(2):532–42. Vutskits L, Gascon E, Tassonyi E, Kiss JZ. Effect of ketamine on dendritic arbor development and survival of immature GABAergic neurons in vitro. Toxicol Sci 2006;91(2):540–9. Wiese C, Rolletschek A, Kania G, Blyszczuk P, Tarasov KV, Tarasova Y, et al. Nestin expression—a property of multi-lineage progenitor cells? Cell Mol Life Sci 2004;61(19–20):2510–22. Wu YY, Bradshaw RA. Effect of nerve growth factor and fibroblast growth factor on PC12 cells: inhibition by orthovanadate. J Cell Biol 1993;121(2):409–22. Yamauchi J, Miyamoto Y, Murabe M, Fujiwara Y, Sanbe A, Fujita Y, et al. Gadd45a, the gene induced by the mood stabilizer valproic acid, regulates neurite outgrowth through JNK and the substrate paxillin in N1E-115 neuroblastoma cells. Exp Cell Res 2007;313(9):1886–96. Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 2005;120(1): 137–49. Yoshimura T, Arimura N, Kaibuchi K. Signaling networks in neuronal polarization. J Neurosci 2006;26(42):10626–30. Yu X, Malenka RC. Beta-catenin is critical for dendritic morphogenesis. Nat Neurosci 2003;6(11):1169–77. Zhang SC, Wernig M, Duncan ID, Brustle O, Thomson JA. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol 2001;19(12):1129–33. Zhang W, Smith A, Liu JP, Cheung NS, Zhou S, Liu K, et al. GSK3beta modulates PACAPinduced neuritogenesis in PC12 cells by acting downstream of Rap1 in a caveolaedependent manner. Cell Signal 2009a;21(2):237–45. Zhang Y, Wang YG, Zhang Q, Liu XJ, Liu X, Jiao L, et al. Interaction of Mint2 with TrkA is involved in regulation of nerve growth factor-induced neurite outgrowth. J Biol Chem 2009b;284(18):12469–79.