Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Nucleic acid analogue wikipedia , lookup
Transposable element wikipedia , lookup
Proteolysis wikipedia , lookup
Bisulfite sequencing wikipedia , lookup
Whole genome sequencing wikipedia , lookup
Zinc finger nuclease wikipedia , lookup
Promoter (genetics) wikipedia , lookup
Biosynthesis wikipedia , lookup
Deoxyribozyme wikipedia , lookup
Gene expression wikipedia , lookup
Multilocus sequence typing wikipedia , lookup
Silencer (genetics) wikipedia , lookup
Genetic code wikipedia , lookup
Community fingerprinting wikipedia , lookup
Genomic library wikipedia , lookup
Protein structure prediction wikipedia , lookup
Endogenous retrovirus wikipedia , lookup
Two-hybrid screening wikipedia , lookup
Non-coding DNA wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
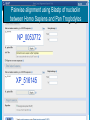
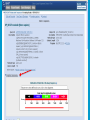
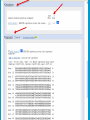
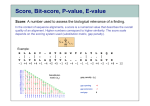
How To Compare Two Sequences? pairwise alignment includes precise tools • Dot plots for graphical analysis • Local or global alignments for residue/residue analysis In the alignment searching different models exist which are designed to encapsulate a variety of characteristics of biological sequences and/or their evolutionary relatedness. Two general model explore aligment: Needleman e Wunsch (1970) considers similarity across the full extent of the sequence (global alignment); Smith e Waterman (1981) focuses on regions of similarity in parts of the sequences (local alignment). 4 steps 1. Definition of the substitution matrix and gap penalties. 2. Alignment of the sequences through a dot matrix. 3. Finding the network with the maximum score also considering gaps. 4. Evaluation of the score derived by the substitution matrix. Basic Local Alignment Search Tool Widely used similarity search tool based on Smith Waterman algorithm Finds best local alignments Provides statistical significance All combinations (DNA/Protein) query and database. • DNA vs DNA • DNA translation vs Protein • Protein vs Protein • Protein vs DNA translation • DNA translation vs DNA translation Many BLAST PROGRAMS . . . BLASTing DNA Sequences Blast Pairwise comparison Running a Blast program NCBI http://blast.ncbi.nlm.nih.gov/Blast.cgi Select the Blast program Write the accession number or cut and paste the query sequence Write the accession number or cut and paste the subject sequence Run the program Pairwise alignment using Blastp of nucleolin between Homo Sapiens and Pan Troglodytes NP_0053772 XP_516145 EXPECTATION VALUE (E-value) IS THE MEASURE MOST COMMONLY USED FOR ESTIMATING SEQUENCE SIMILARITY IS ASSOCIATED WITH A SPECIFIC SCORE (“S”) AND SEQUENCE IS THE NUMBER OF DIFFERENT ALIGNMENTS WITH SCORES EQUIVALENT TO OR BETTER THAN “S”, THAT ARE EXPECTED TO OCCUR BY CHANCE. THE E-VALUE THE LOWER THE E VALUE, THE MORE SIGNIFICANT THE SCORE. IF A MATCH IS HIGHLY UNEXPECTED, IT PROBABLY RESULTS FROM SOMETHING OTHER THAN CHANCE COMMON ORIGIN IS THE MOST LIKELY EXPLANATION E-value An E-value of 1 assigned to a hit can mean that in the database, one might expect to see one match with a similar score simply by chance. This means that the lower the E-value, or the closer it is to “0”, the higher is the “significance” of the match. However, it is important to note that searches with short sequences can be virtually identical and have relatively high Evalue. This is because the calculation of the E-value also takes into account the length of the query sequence. This is because shorter sequences have a high probability of occurring in the database purely by chance. Meaning for E-Values Low E-value good hit • 1 = bad e-Value • 10-3 = borderline E-value • 10-4 = good E-value • 10-10 = very good E-value E-values lower than 10-4 indicate possible homology E-values higher than 10-4 require extra evidence to support homology BLAST Output: Alignments Identical match positive score (conservative) gap Negative or zero note A pairwise sequence alignment from a BLAST report The alignment is preceded by the sequence identifier, the full definition line, and the length of the matched sequence, in amino acids. Next comes the bit score (the raw score is in parentheses) and then the E-value. The following line contains information on the number of identical residues in this alignment (Identities), the number of conservative substitutions (Positives), and if applicable, the number of gaps in the alignment. Finally, the actual alignment is shown, with the query on top, and the database match is labeled as Sbjct, below. The numbers at left and right refer to the position in the amino acid sequence. One or more dashes (–) within a sequence indicate insertions or deletions. Amino acid residues in the query sequence that have been masked because of low complexity are replaced by Xs (see, for example, the fourth and last blocks). The line between the two sequences indicates the similarities between the sequences. If the query and the subject have the same amino acid at a given location, the residue itself is shown. Conservative substitutions, as judged by the substitution matrix, are indicated with +. The Alignments Look for clusters of identity Gray residues are low-complexity regions Grayed-out regions have been removed from your sequence to avoid false hits Pair-wise comparison between Human nucleolin NP_005372.2 and chimpanzee nucleolin XP_516145.3 Search for similarity of albumin between human and chimpanzee human and Mus musculus human and Gallus gallus Link rete Similarity searching in the protein data base BLAST MAKES PAIRWISE COMPARISON OF THE QUERY SEQUENCE TO EACH SEQUENCE IN THE DATABASE PROCEDURE • SEARCH FOR LOCAL ALIGNMENT • EVALUATE THE SCORE • SAVE THE HIGH SCORES • REPEAT THE STEPS FOR EACH SEQUENCE Running blastp http://blast.ncbi.nlm.nih.gov/Blast.cgi Select a database to search Write the accession number or cut and paste the query sequence Click the BLAST button Reading BLAST Output Graphic Display • Overview of the alignments Hit List • Gives the score of each match Alignments • Details of each alignment The Graphic Display The Horizontal Axis (0-700) corresponds to your protein (query) Color codes indicate match’s quality • Red: very good • Green: acceptable • Black: bad Thin lines join independent matches on the same sequence The Hit List Sequence accession number • Depends on the database Description • Taken from the database Bit score • High bit score = good match E-Value • Low E-value = good match Links • • Genome Uniref, database of transcripts BLAST Output: Descriptions Sorted by e values 5 X 10-14 Link to entrez Gene Linkout NUCLEOLIN GENE IN HOMO SAPIENS Search for homologous sequences To human nucleolin In refseq protein database In mammals Exclude model proteins Use the default algorithm parameters output results in Recen results window Basic BLAST: Nucleotide BLAST and BLAST-like programs Megablast nucleotide only • Contiguous megablast • Nearly identical sequences • Discontiguous megablast • Cross-species comparison Nucleotide Databases: Human and Mouse Human and mouse genomic and transcript now default Separate sections in output for mRNA and genomic Direct links to Map Viewer for genomic sequences Nucleotide Databases: Traditional blastn tblastn tblastx Universal Form: Nucleotide Less Sensitivity More More Speed Less Search for homologous sequences To human nucleolin transcript In human genomic + transcript Exclude model proteins Select program for highly similar sequences Use the default algorithm parameters output results in Recen results window Link to Genome View Total Score: All Segments Sortable Results Separate Sections for Transcript and Genome Link to Map Viewer Genome BLAST Genome BLAST pages Map Viewer Homepage Search for homologous sequences to human nucleolin mRNA RefSeq in Bos Taurus reference assembly