Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
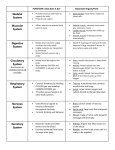
Molecular basis of diseases-‐ muscles atrophy Muscular System Functions • • • • • • • • • • Body movement (Locomotion) Maintenance of posture Respiration Diaphragm and intercostal contractions Communication (Verbal and Facial) Constriction of organs and vessels Peristalsis of intestinal tract Vasoconstriction of b.v. and other structures (pupils) Heart beat Production of body heat (Thermogenesis) Types of Muscle • • • Skeletal – Attached to bones – Makes up 40% of body weight – Responsible for locomotion, facial expressions, posture, respiratory movements, other types of body movement – Voluntary in action; controlled by somatic motor neurons Smooth – In the walls of hollow organs, blood vessels, eye, glands, uterus, skin – Some functions: propel urine, mix food in digestive tract, dilating/constricting pupils, regulating blood flow, – In some locations, autorhythmic – Controlled involuntarily by endocrine and autonomic nervous systems Cardiac – Heart: major source of movement of blood – Autorhythmic – Controlled involuntarily by endocrine and autonomic nervous systems Muscles disorders § Muscle cramp: sustained painful contraction – hyperexcitability of the motor unit, countered with stretching § Overuse – excessive use that causes tearing in the muscle structures (fibers, sheaths, tendon connection) § Disuse- loss of muscle activity causes muscle atrophy because of loss of blood flow, can recover is disuse is less than a year § Acquired disorders – infectious diseases and toxin poisoning that lead to muscle weakness or paralysis § Inherited disorders - § Duchenne’s muscular dystrophy – muscle degenrates from pelvis up, happens most often in women, people live to be 20-30, die of respiratory failure § Dystrophin –links actin to proteins in cell membrane § McArdle’s disease – limited exercise tolerance Glycogen to glucose-6-phosphate – enzyme missing thus muscles do not have the energy source available Muscles atrophy Atrophy is defined as a decrease in the size of a tissue or organ due to cellular shrinkage; the decrease in cell size is caused by the loss of organelles, cytoplasm and proteins. It can be caused by immobilization or loss of neural stimulation. Video Causes of Muscle Atrophy Unused muscles can waste away if you are not active. However, this takes time. Even after it begins, this type of atrophy can often be reversed with exercise and improved nutrition. Muscle atrophy can also happen if you are bed-ridden or unable to move certain body parts due to a medical condition. Astronauts are subject to some muscle atrophy after a few days of weightlessness. Other causes for muscle atrophy include: • • lack of physical activity (for any reason) aging • • • • • • • alcohol-associated myopathy (pain and weakness in muscles due to excessive drinking over long periods of time) burns injuries and broken bones malnutrition spinal cord injuries stroke long-term corticosteroid therapy Diseases can cause muscles to waste away or can make movement difficult, leading to muscle atrophy. These include: • • • • • • • • • • • amyotrophic lateral sclerosis (ALS, also known as Lou Gehrig’s disease), which affects nerve cells that control voluntary muscle movement dermatomyositis (a muscle disease) Guillain-Barre syndrome (an autoimmune disease that leads to nerve inflammation and muscle weakness) multiple sclerosis (MS, an autoimmune disease that can make it difficult to move) muscular dystrophy (an inherited disease that causes muscle weakness) neuropathy (damage to a nerve or nerve group, resulting in loss of sensation or function) osteoarthritis (the most common form of arthritis; causes reduced motion in the joints) polio (a viral disease affecting muscle tissue that can lead to paralysis) polymyositis (an inflammatory disease) rheumatoid arthritis (an autoimmune disease) spinal muscular atrophy (SMA, a hereditary disease causing arm and leg muscles to waste away) Signs of Muscle Atrophy You may have muscle atrophy if: • • • one of your arms or legs is noticeably smaller than the other you are experiencing marked weakness in one limb you have been physically inactive How Muscle Atrophy Is Diagnosed Your doctor will take a complete medical history and to understand all of your symptoms. Tell him or her about old or recent injuries you’ve experienced and previously diagnosed medical conditions. List prescriptions, over-the counter medications, and supplements you are taking and your symptoms. Your doctor may order additional tests to help with the diagnosis and to rule out certain diseases. These tests may include: • • • • • • • blood tests X-rays magnetic resonance imaging (MRI) computed tomography (CT) scan nerve conduction studies muscle or nerve biopsy electromyography (EMG) This Review discusses the latest findings and emerging concepts related to pathways controlling muscle atrophy in physiological and pathological conditions. In particular, we focus on the ubiquitin-‐proteasome machinery and the autophagy-‐lysosome machinery, the two most important cell proteolytic systems that control protein turnover in muscle. The involvement of these systems in muscle physiopathology, as well as the signalling pathways controlling their activity, have been unravelled only in recent years, and evidence indicates that these two processes play a pivotal role in regulating overall muscle homeostasis. Molecular base of muscle atrophy Figure 1 illustrates the organizational hierarchy of molecular components that may be involved in the atrophy pathway. Molecular triggers and signaling molecules involved in muscle atrophy Role of Decreased Protein Synthesis in Disuse Atrophy 01. Myostatin A protein belonging to the TGF-β family, known as myostatin, has been shown to be a strong negative regulator of muscle growth. Knockout or mutation of this protein produces animals with markedly enlarged muscles as a result of hypertrophy and hyperplasia. Conversely, myostatin can induce atrophy via an inhibitory effect on translation. Systemic administration of this negative growth regulator leads to muscle wasting in mice, and treatment of cultured muscle cells with recombinant myostatin has resulted in the loss of protein and reduced protein synthesis rates. Moreover, myostatin expression is increased in some types of muscle atrophy. Human immune deficiency virus (HIV)-infected men have shown higher levels of serum myostatin , indicating that myostatin may contribute to cachexia-type atrophy. 02. Glucocorticoids The synthetic glucocorticoid dexamethasone is widely used to induce muscle proteolysis either in vivo or in cell culture. In skeletal muscle; glucocorticoids decrease the rate of protein synthesis and increase the rate of protein degradation. Both disuse atrophy and cachexia are associated with increases in circulating glucocorticoid levels. Glucocorticoids (GCs) are a class of steroid hormones that bind to the glucocorticoid receptor (GR), which is present in almost every vertebrate animal cell. The name glucocorticoid (glucose + cortex + steroid) derives from its role in the regulation of the metabolism of glucose, its synthesis in the adrenal cortex, and its steroidal structure. A less common synonym is glucocorticosteroid. Moreover, the binding capacity of corticosteriods also was increased markedly with disuse atrophy. However, when adrenalectomized animals (Adrenalectomy is the surgical removal of one or both (bilateral adrenalectomy) adrenal glands) underwent unloading, with or without cortisol treatment, atrophy still occurred .Importantly, treatment of unloaded rats with an inhibitor of glucocorticoids, RU-38486, also did not inhibit disuse atrophy. Thus glucocorticoids do not appear to be required for disuse atrophy. In the case of cachexia, glucocorticoids seem to be a contributing factor to muscle wasting in part because rats treated with RU-38486 plus TNF-α showed reduced proteolysis, but protein loss was not completely attenuated. TNF-‐α and Other Cytokines There is no evidence that TNF-α or other cytokines are involved in disuse atrophy. However, there is significant literature on the role of cytokines in cachexia showing that TNF-α and other cytokines such as IL-1 and IL-6 are increased in these conditions. Administration of TNF-α can induce cachexia, and blockade of TNF-α by torbafylline in rats with either cancer or sepsis prevents muscle wasting . TNF-α treatment alone also leads to increased protein degradation in cultured muscle cells. In addition to TNF-α, a protein named PIF (proteolysis-inducing factor) also has been shown to have the potential for acting as a trigger of atrophy in cancer cachexia. Although isolated from more than one tumor type, this molecule is only in the very early stages of study, but its activity has been linked to NF-κB activation. To date, there have been no published studies on the potential role of PIF in disuse atrophy. NF-κB Signaling NF-κB is a family dimeric proteins encoded by five gene members: RelA/p65, RelB, c-Rel, NF-κB1/p50, and NF-κB2/p52 (the last 2 of which derive from precursor subunits, p105 and p100, respectively). 2 path ways-Canonical & non canonical pathway Oxidative Stress Another area that has received some degree of attention is the generation of reactive oxygen species (ROS) in muscle atrophy. Unloading atrophy results in upregulation of Cu, Zn superoxide dismutase and is more damaging because of a concomitant decrease of catalase, glutathione peroxidase, and, possibly, Mn superoxide dismutase (56), the systems that would normally act to metabolize increases in ROS. Treatment of muscle cells with H2O2 leads to increased protein breakdown, decreased myosin expression, and increased expression of components of the ubiquitin-proteasome proteolytic pathway. Role of Increased Proteolysis in Muscle Atrophy There is more know about the role of increased proteolysis than there is of decreased synthesis in disuse atrophy. At least half of total muscle protein is myofibrillar protein, and this fraction is lost at a faster rate than other muscle proteins during atrophy. Contributions of three major proteolytic systems to skeletal muscle protein loss: the cytosolic calcium-‐dependent calpain system, the lysosomal proteases (i.e., cathepsins), and the ATP-‐dependent ubiquitin-‐proteasome system. Role of calpains in disuse atrophy It has been known for some time that the calpains are unable to degrade actin and myosin, although they have activity at a few specific sites. Proteins that are involved in the assembly and scaffolding of myofibrillar proteins such as titin, vinculin, C-‐protein, nebulin, and others are known calpain substrates. Role of lysosomal proteolysis in disuse atrophy It has shown increases in various isoforms of cathepsin mRNAs in disuse atrophy. However, when atrophying muscle resulting from disuse is treated with agents that block lysosomal acidification (96) or with agents that directly inhibit cathepsins myofibrillar protein degradation rates are not significantly affected and total protein degradation rates are only slightly reduced. Role of the ubiquitin-‐proteasome system in disuse atrophy In muscle, the ubiquitin-proteasome system is required to remove sarcomeric proteins upon changes in muscle activity. A decrease in muscle mass is associated with: (1) Increased conjugation of ubiquitin to muscle proteins (2) Increased proteasomal ATP-dependent activity (3) Increased protein breakdown that can be efficiently blocked by proteasome inhibitors and (4) Upregulation of transcripts encoding ubiquitin, some ubiquitin-conjugating enzymes (E2), a few ubiquitin-protein ligases (E3) and several proteasome subunits. Mechanisms E1 enzymes activate ubiquitin proteins after the cleavage of ATP. The ubiquitin is then moved from E1 to members of the E2 enzyme class. The final ubiquitylation reaction is catalyzed by members of the E3 enzyme class. E3 binds to E2 and the protein substrate, inducing the transfer of ubiquitin from E2 to the substrate. Once the substrate is polyubiquitylated, it is docked to the proteasome for degradation. Note that polyubiquitin chains can be removed by de-‐ubiquitylating enzymes [ubiquitin-‐specific processing proteases (USPs)]. The components of this system that contribute to muscle wasting are depicted. ZNF216 is involved in the recognition and delivery to the proteasome of ubiquitylated proteins during muscle atrophy. IRS1, insulin receptor substrate 1; Ub, ubiquitin. Atrogin-‐1 regulates the half-‐life of the MyoD transcription factor and of eIF3f, which is crucial for protein synthesis. Fbxo40 regulates the half-‐life of IRS1, an essential factor for IGF1/insulin signalling Whereas MuRF1 regulates the half-‐life of several sarcomeric proteins. E3 ubiquitin ligases are depicted in green, with arrows pointing to their substrates. Comparing gene expression in different models of muscle atrophy led to the identification of a subset of genes that are commonly up-‐ or down regulated in atrophying muscle. Genes are believed to regulate the loss of muscle components and are called atrophy-‐related genes or ‘atrogenes’ Together, these findings revealed that muscle atrophy is an active process controlled by specific signalling pathways and transcriptional programs. Furthermore, the genes induced most strongly were found to encode two muscle-‐ specific ubiquitin ligases, atrogin-‐1 (also known as MAFbx) and MuRF1. Valuable information on the role of specific components of the ubiquitin-‐ proteasome system in muscle was obtained by generating genetically modified animals .Mice lacking atrogin-‐1 and MuRF1 are resistant to muscle atrophy induced by denervation. Moreover, knockdown of atrogin-‐1 prevents muscle loss during fasting, whereas MuRF1 knockout mice (but not atrogin-‐1 knockout mice) are resistant to dexamethasone-‐induced muscle atrophy. However, only a few muscle proteins have been identified as substrates for atrogin-‐1 thus far, and they all seem to be involved in growth-‐related processes or survival pathways. Atrogin-‐1 promotes degradation of MyoD (MyoD is a protein with a key role in regulating muscle differentiation. MyoD belongs to a family of proteins known as myogenic regulatory factors (MRFs)), a key muscle transcription factor, and of eIF3f, an important activator of protein synthesis. MuRF1 was reported to interact and control the half-‐life of important muscle structural proteins, including troponin I , myosin heavy chains, myosin binding protein C and myosin light chain. A recent paper reported Trim32 as a crucial E3 ligase for the degradation of thin filaments (actin, tropomyosin and troponins), α-‐actinin and desmin. However, Trim32 knockout mice are not protected from atrophy, but show impairment in the recovery of muscle mass after atrophy. Summary Skeletal muscle atrophy attributable to muscular inactivity has significant adverse functional consequences. While the initiating physiological event leading to atrophy seems to be the loss of muscle tension and a good deal of the physiology of muscle atrophy has been characterized, little is known about the triggers or the molecular signaling events underlying this process. Decreases in protein synthesis and increases in protein degradation both have been shown to contribute to muscle protein loss due to disuse, and recent work has delineated elements of both synthetic and proteolytic processes underlying muscle atrophy. It is also becoming evident that interactions among known proteolytic pathways (ubiquitin-‐proteasome, lysosomal, and calpain) are involved in muscle proteolysis during atrophy. Factors such as TNF-‐α, glucocorticoids, myostatin, and reactive oxygen species can induce muscle protein loss under specified conditions. Also, it is now apparent that the transcription factor NF-‐κB is a key intracellular signal transducer in disuse atrophy. Transcriptional profiles of atrophying muscle show both up-‐ and downregulation of various genes over time, thus providing further evidence that there are multiple concurrent processes involved in muscle atrophy. The purpose of this review is to synthesize our current understanding of the molecular regulation of muscle atrophy. Spinal muscular atrophy Spinal muscular atrophy (SMA) is an autosomal recessive disorder that is one of the most common genetic causes of childhood mortality. The main characteristic of the disease is progressive loss of spinal cord motor neurons, resulting in skeletal muscle denervation with subsequent weakness, atrophy, and paralysis of voluntary muscles. Disease caused by low levels of SMN proteins. SMN (survival motor neuron) protein is found throughout the body, with high levels in the spinal cord. This protein is particularly important for the maintenance of specialized nerve cells called motor neurons, which are located in the spinal cord and the part of the brain that is connected to the spinal cord (the brainstem). Motor neurons control muscle movement.SMN proteins encoded by SMN genes. SMN1 gene location Cytogenetic Location: 5q13.2 Molecular Location on chromosome 5: base pairs 70,924,940 to 70,953,011 The SMN1 gene is located on the long (q) arm of chromosome 5 at position 13.2. A small amount of SMN protein is produced from a gene similar to SMN1 called SMN2. The SMN2 gene provides instructions for making several versions of the SMN protein, but only one version is functional; the other versions are smaller and easily broken down. A strong correlation between the loss of motor neurons and the reduction of nuclear staining for SM-containing snRNPs in mouse models of SMA strongly suggests that the SMN deficiency causes disease by a defect in pre-mRNA splicing. Trans effects: mutations that affect the basal splicing machinery (classical splicing methods) . About 95 percent of individuals with spinal muscular atrophy have mutations that delete a section called exon 7 in both copies of the SMN1 gene in each cell. As a result, little or no SMN protein is made. In about 5 percent of people with this disorder, one copy of the SMN1 gene has a deletion of exon 7, and the other copy has a different mutation that disrupts the production or function of the SMN protein. Researchers have identified at least 65 mutations in the SMN1 gene that cause spinal muscular atrophy. Despite the potential to encode the identical protein, the SMN2 gene does not completely compensate for loss of SMN1 function because one of the nucleotide substitutions disrupts an ESE in exon 7 that causes the exon to be skipped. Motor neurons seem to be particularly vulnerable to a shortage of the SMN protein and die prematurely. Researchers suggest that a shortage of SMN protein leads to the inefficient assembly of the machinery needed to process pre-mRNA. Without mature mRNA, the production of proteins necessary for cell growth and function is disrupted. Some research findings indicate that a shortage of SMN protein impairs the formation and function of axons and dendrites, possibly leading to the death of neurons. While the cause of neuronal death is unclear, it is the loss of motor neurons that leads to the signs and symptoms of spinal muscular atrophy. The resulting SMN2 ΔE7 mRNA encodes a truncated protein missing the Cterminal 16 residues and is thought to be nonfunctional. The best characterized role for the SMN complex is in the assembly of U1, U2, U4, and U5 snRNPs. Unlike humans, mice have only one Smn gene. Smn −/− mice die at the blastocyst stage, and Smn +/−mice develop symptoms strikingly similar to SMA. Smn +/− mice are normal at birth but develop SMA-like symptoms within days owing to a normal developmentally regulated decline in which SMN protein levels in the spinal cord drop to <50% of fetal levels, primarily between postnatal days 5 and 15. In some cases of spinal muscular atrophy, in addition to their SMN1 gene mutations, affected individuals have three or more copies of the SMN2 gene in each cell. Extra SMN2 genes can help replace some of the SMN protein that is lost due to mutations in the SMN1 genes. In general, symptoms are less severe and begin later in life in affected individuals with three or more copies of the SMN2 gene compared with those who have two copies of the gene. These results indicate that postnatal motor neurons require higher steady-state levels of SMN protein than other metabolically active tissues. Trans effects: mutations that affect regulators of alternative splicing Myotonic dystrophy Myotonic dystrophy (DM) is the one human disease in which disease phenotype has been directly linked to disrupted regulation of alternative splicing.DM is an autosomal dominant disorder and the most common form of adult-‐onset muscular dystrophy, with a worldwide incidence of 1 in 8000. DM is unusual because of its phenotypic variability even within families and the diversity of tissues affected. Symptoms include skeletal muscle hyperexcitability (myotonia), progressive muscle wasting, cardiac conduction defects, cataracts, smooth muscle dysfunction, testicular atrophy, an unusual form of insulin resistance, and neuropsychiatric and cognitive disturbances. Two types of DM have been identified. The most common form is type 1 (DM1), which is caused by a CTG expansion in the 3′ untranslated region (UTR) of the DM protein kinase (DMPK) gene located on Chromosome 19q13.3. Disease severity and age of onset correlate with repeat length, which ranges from 80 to thousands of repeats. Unaffected individuals have fewer than ∼40 repeats. DM type 2 (DM2) is caused by a large CCTG expansion in intron 1 of the ZNF9 gene on Chromosome 3q21.