Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Adaptive immune system wikipedia , lookup
Polyclonal B cell response wikipedia , lookup
Molecular mimicry wikipedia , lookup
Innate immune system wikipedia , lookup
Hygiene hypothesis wikipedia , lookup
Multiple sclerosis signs and symptoms wikipedia , lookup
Cancer immunotherapy wikipedia , lookup
Sjögren syndrome wikipedia , lookup
Adoptive cell transfer wikipedia , lookup
Psychoneuroimmunology wikipedia , lookup
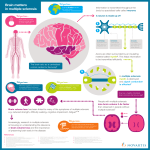
Pathophysiology of multiple sclerosis wikipedia , lookup
Immunosuppressive drug wikipedia , lookup
Immune activation in multiple sclerosis and interferon- therapy PhD Thesis Martin Krakauer, MD. University of Copenhagen Submitted 31 August 2006 Defended 25 September 2007 p2 Abbreviations APC Antigen Presenting Cell BBB Blood-Brain Barrier CD Cluster of Differentiation CNS Central Nervous System EAE Experimental Autoimmune Encephalomyelitis ELISA Enzyme-Linked Immunosorbent Assay HLA Human Leukocyte Antigen IFN Interferon IL Interleukin MHC Major Histocompatibility Complex MMP Matrix MetalloProteinase MRI Magnetic Resonance Imaging mRNA Messenger RNA (RiboNucleic Acid) MS Multiple Sclerosis PBMC Peripheral Blood Mononuclear Cell PCR Polymerase Chain Reaction PPMS Primary Progressive Multiple Sclerosis RRMS Relapsing-Remitting Multiple Sclerosis SPMS Secondary Progressive Multiple Sclerosis Th T helper type lymphocyte (CD4+ T cell) TIMP-1 Tissue inhibitor of metalloproteinase 1 TNF Tumour Necrosis Factor TRAIL Tumour necrosis factor-Related Apoptosis Inducing Ligand VCAM Vascular Cell Adhesion Molecule p3 Contents Abbreviations ............................................................................................................................ 2 Contents ..................................................................................................................................... 3 1. Introduction (incl. publications and acknowledgements) ..................................................... 4 1.1. Papers included in the PhD thesis ......................................................... 5 2. Theoretical section ................................................................................................................ 6 2.1. Multiple sclerosis: epidemiology and clinical features ......................... 6 2.2. Immunology and pathophysiology of multiple sclerosis ...................... 7 2.3. Immunomodulatory treatments for multiple sclerosis ........................ 11 2.4. Biomarkers .......................................................................................... 13 2.4.1 Type 0 biomarkers in multiple sclerosis ............................................. 14 2.4.2 Type 1 biomarkers in multiple sclerosis ............................................. 16 3. Aim of the thesis ................................................................................................................. 18 4. Summary of own studies (brief materials, methods, and results) ....................................... 19 4.1. P-I: CD26 paper .................................................................................. 19 4.2. P-II: Chemokine paper ........................................................................ 19 4.3. P-III: Cytokine paper ........................................................................... 20 4.4. Overall conclusions of own studies ..................................................... 21 5. Critique of materials and methods ...................................................................................... 22 5.1. General methodological considerations .............................................. 22 5.2. Technical considerations ..................................................................... 23 6. Discussion ........................................................................................................................... 26 6.1. Immunological characteristics of MS patients .................................... 26 6.2. Immunological effects of IFN- therapy ............................................ 27 6.3. Putative biomarkers in MS and IFN- therapy ................................... 30 7. Future perspectives .............................................................................................................. 31 8. References ........................................................................................................................... 32 9. Summary ............................................................................................................................. 43 10. Summary in Danish / Dansk resumé ................................................................................. 45 11. Appendices ........................................................................................................................ 47 9.1. P-I: CD26 paper 9.2. P-II: Chemokine paper 9.3. P-III: Cytokine paper p4 1. Introduction The work forming the basis of this PhD thesis was performed during my employment at the Danish Multiple Sclerosis Research Center, Department of Neurology, Copenhagen University Hospital Rigshospitalet, Denmark / University of Copenhagen, in the period from 2003 to 2006. The thesis is based on three manuscripts, P-I, P-II, and P-III, of which P-II has been published, and P-I and P-III have been submitted for publication. All manuscripts address aspects of the immunology in multiple sclerosis, and the immunological effects of treatment with interferon-. I wish to thank Dr. Finn Sellebjerg and Professor Per Soelberg Sørensen for their invaluable help and support during all phases of my research projects. I especially want to thank Finn Sellebjerg for sharing with me the impressive amount of knowledge and expertise that he holds, as well as for providing an inspiring, friendly, and informal work environment. I also want to thank the laboratory staff at the Neuroimmunology Laboratory at Copenhagen University Hospital, Rigshospitalet. Thanks are due to Susanne Velgaard, Henriette Egeblad, Anne Marie Nordvig Petersen, and Rikke Kroager without whom I would not have been able to carry out all the analyses included in the thesis. I am also grateful to Dr. Poul Erik Hyldgaard Jensen for his help in the laboratory and the nurses in the MS clinic who have helped me recruit patients for the studies. Finally, I wish to express my gratitude to all the patients that have participated in the studies. Without their help, no clinical research would be possible. It is my hope that my work has made a contribution, albeit small, to the future development of more efficacious treatments for multiple sclerosis. p5 1.1. Papers included in the PhD thesis. P-I: (CD26 Paper) Krakauer M, Sorensen PS, Sellebjerg F. CD4+ memory T cells with high CD26 surface expression are enriched for Th1 markers and correlate with clinical severity of multiple sclerosis. J Neuroimmunol 2006; 181: 157-164. P-II: (Chemokine Paper) Krakauer M, Sorensen PS, Khademi M, Olsson T, Sellebjerg F. Dynamic T-lymphocyte Chemokine Receptor Expression Induced by Interferon-beta Therapy in Multiple Sclerosis. Scand J Immunol 2006; 64 (2): 155-163. P-III: (Cytokine Paper) Krakauer M, Sorensen PS, Khademi M, Olsson T, Sellebjerg F. Increased IL-10 mRNA and IL-23 mRNA expression in multiple sclerosis. Interferon-β treatment increases IL-10 mRNA expression while reducing IL-23 mRNA expression. Submitted. p6 2. Theoretical section 2.1. Multiple sclerosis: epidemiology and clinical features Multiple sclerosis (MS) is a demyelinating disease of the CNS. Affecting approximately one in 1000 in high-risk areas such as Denmark, it is a leading cause of disability in younger adults [Koch-Henriksen et al. 1992; Ebers and Sadovnick 1993]. The age of onset is typically between twenty and forty years, and the life expectancy of MS patients is 5-10 years shorter than that of the general population [Brønnum-Hansen et al. 2004]. About two thirds of MS patients are women. Geographically, incidence rates increase with the distance from the Equator, but ethnicity also contributes. MS has heterogeneous clinical presentations: about 85-90 % present with a relapsingremitting form (RRMS) while 10-15 % have primary progressive MS, or PPMS [Lublin and Reingold 1996]. After 10-15 years, RRMS often develops into a secondary progressive form (SPMS), characterised by progressive loss of neurological functions, that resembles PPMS, with or without superimposed relapses [Confavreux and Vukusic 2006]. RRMS is characterised by episodes of acute relapses of focal inflammation in the brain or spinal cord white matter with demyelination and some degree of axonal and neuronal loss. Lesions can be clinically silent depending on their location and size, but often produce corresponding neurological symptoms, defining an acute MS attack. Consequently, symptoms are variable and include limb paresis, impaired vision, paraesthesias, diplopia, vertigo, bladder symptoms, cognitive impairment, and fatigue. The acute MS attack generally remits within a few weeks or months, leaving behind varying degrees of residual symptoms from the affected area of the CNS. The average relapse-rate in untreated RRMS patients is approximately 0.5 per year [Confavreux and Compston 2005]. Recovery from relapses is variable. In the early stages of MS, recovery is often complete or near complete, probably due to the plasticity and reserve capacity of the brain. In the later stages of MS, recovery is often incomplete as the reserve capacity of the brain is declining due to the progressive neuronal loss [Pantano et al. 2005; Cooke and Bliss 2006]. p7 2.2. Immunology and pathophysiology of multiple sclerosis Although the cause of MS is unknown, epidemiological and genetic studies support the concept of a multi-factorial aetiology where environmental factors trigger a chronic inflammation in a genetically susceptible person. The histopathological hallmark of MS is plaques of focal CNS inflammation, predominantly located in periventricular white matter around a central blood vessel. The inflammatory cells mainly comprise macrophages and Tlymphocytes (T cells) although B-lymphocytes and plasma cells are also found in the perivascular space. The discovery of prominent grey matter and more diffuse white matter inflammation, at least in some MS patients, has recently added to the complexity of MS pathology [Kutzelnigg et al. 2005]. Furthermore, data support the existence of distinct interindividual morphological heterogeneity of the white matter plaques [Lucchinetti et al. 2000]. Although never formally proven, the inflammation is believed to involve an autoimmune reaction directed against CNS antigens, primarily myelin components. Support for this notion comes from animal studies in which an MS-like disease, experimental autoimmune encephalomyelitis (EAE), can be induced by immunising susceptible animal strains, predominantly mice and rats, with CNS antigens in combination with an adjuvant. The immunisation results in the formation of demyelinating plaques in the CNS, with histopathological similarities to MS, and ensuing ascending paralysis. Support for the autoimmune hypothesis in MS also comes from genetic studies showing an increased risk of developing MS in individuals with the HLA-DR2 haplotype belonging to the MHC class II tissue type molecule (Odds Ratio 3-4 for heterozygotic carriers, 8 for homozygotic carriers) [Svejgaard et al. 1983; Modin et al. 2004; Oksenberg et al. 2004]. This molecule is required for the presentation of antigens to T cells by antigen-presenting cells (APCs) and is therefore closely linked to the functions of the immune system. Indeed, a triple transgenic humanised mouse model carrying human DR2, a myelin basic protein (MBP)-specific T cell receptor (TCR) and CD4, develops spontaneous disease under certain experimental conditions [Madsen et al. 1999]. It is believed that the initial formation of the inflammatory CNS plaque is preceded by systemic activation of myelin-reactive CD4+ T helper (Th) cells through interaction with p8 the antigen/MHC class II-complex of the APCs. Upon activation, these cells acquire a phenotype that allows them to migrate into the CNS, an area inaccessible to resting immune cells. In the CNS the Th cells are reactivated upon encounter with their cognate antigen presented by MHC class II molecules on perivascular cells, a CNS-resident cell type with phagocytic and antigen-presentation capabilities. This secondary activation triggers the release of proinflammatory cytokines, chemokines, and other mediators, in turn attracting and activating other immune effector cells (CD8+ T cells, B cells, and microglia / macrophages). These cells establish the inflammatory lesion and cause demyelination and axonal damage though cytotoxicity, antibody opsonisation, and complement-mediated membrane attack (fig 1). Not only acting as attractors and activators of other immune effector cells, it has recently become clear that a subset of CD4+ Th cells (CD4+ CD28-) themselves also exhibit cytotoxic capabilities [Appay 2004; Amyes et al. 2005]. Figure 1. Proposed mechanism for the pathogenesis of the MS inflammatory plaque formation. (Adapted from Holmes et al, Expert reviews in molecular medicine © 2005 Cambridge University Press). p9 Although there is controversy regarding which cell type is mainly responsible for the formation of the CNS inflammatory lesion, the Th cells (CD4+ T cells) are often suggested. There are two major arguments supporting a central role for Th cells: 1) Most EAE models can be induced by transferring activated myelin-reactive Th cells from a diseased animal into the blood stream of an unaffected animal, a process termed adoptive transfer. In contrast, few EAE models are inducible by adoptive transfer of antigen-specific CD8+ T cells [Huseby et al. 2001; Sun et al. 2001; Ford and Evavold 2005]. 2) Th cells are presented to their cognate antigen by the APCs via the MHC class II tissue type molecule while CD8+ T cells bind antigen peptides bound to MHC class I molecules. As genetic linkage studies show an increased risk of acquiring MS in individuals with the MHC class II haplotype HLA-DR2, this indicates a crucial role of APC-Th cell interactions in MS pathogenesis. Being involved in CD8+ T cell antigen presentation, MHC class I polymorphisms have not been associated as strongly with MS susceptibility [Harbo et al. 2004]. Healthy individuals as well as MS patients have systemically circulating myelinreactive T cells, but in MS patients the cells are more activated, and they are only found intrathecally in MS patients [Zhang et al. 1994; Lovett-Racke et al. 1998; Burns et al. 1999; Bielekova et al. 2004]. The question is how the myelin-reactive T cells become activated in MS patients. Several models have been proposed, and some of them are listed here (reviewed in [Fujinami et al. 2006]). Molecular mimicry: The organism is infected with a virus that encodes proteins that share an immunologic epitope with a myelin protein, thus inadvertently activating T cells that recognise myelin autoantigens [Wucherpfennig 2001]. Super-antigens: Some viral or bacterial so-called super-antigens are capable of cross-linking the MHC class II molecule on the APC with the T cell receptor complex, resulting in polyclonal T cell activation, irrespective of the specificity of the T cell receptor [Wucherpfennig 2001]. Bystander activation: During an unspecific infection APCs become activated. The activation threshold of a pre-primed myelin-reactive T cell located in close proximity to such an APC can be lowered due to the release of pro-inflammatory cytokines from the APC [Banerjee et al. 2005]. Dual T cell receptors: Counter to previous belief, some T cells express dual T cell receptors with differing specificities. Theoretically, if one of these receptors recognises a viral antigen, infection with this virus could lead to clonal expansion of the T cell and, p 10 consequently, enhanced immune responses towards epitopes recognised by the other receptor, e.g. from a myelin peptide [He et al. 2002]. None of these models has yet been proven responsible for the pathogenesis of MS. The phenotype of the encephalitogenic Th cells in EAE is that of T helper type 1 cells (Th1) that secrete interferon (IFN)- and tumour necrosis factor (TNF, previously termed TNF-). Th1 cells are also increased in active MS [Correale et al. 1995]. These cells are generally mobilised in response to intracellular viral pathogens and bacterial infections, causing activation and attraction of phagocytes, and B lymphocyte IgG isotype shifts promoting opsonisation of bacteria. The counterparts to Th1 cells are the Th2 cells, which are characterised by secretion of interleukin (IL)-4, IL-5, and IL-13. Th2 cells deviate the immune response towards defence against extracellular parasitic infections through IgE production and mast/eosinophil degranulation (IL-10 belongs to the Th2 cytokines in mice, but in humans many T cells, including Th1 and Th2 cells, can secrete IL-10 [Delprete et al. 1993]). Th1 and Th2 cells are to a certain degree mutually antagonistic as Th1 cytokines inhibit Th2 responses and vice versa. Thus, the Th cell acts as a key orchestrator of the adaptive immune responses. In general, many autoimmune diseases (type I diabetes, rheumatoid arthritis, inflammatory bowel disease, etc.) are associated with a Th1 deviation whereas allergic conditions such as atopy and asthma are associated with a Th2 pattern. The differentiation into Th1 cells or Th2 cells are controlled through the action of several transcription factors. Expression of Tbet directs the Th cell toward a Th1 phenotype whereas expression of GATA-3 promotes Th2 differentiation [Murphy and Reiner 2002]. In EAE, genetic knockout of IL-12, a potent APC-derived inducer of Th1 deviation, prevents clinical disease, supporting the role of a Th1 deviation in the immunopathogenesis of EAE. However, in these experiments the knockout mice were only deficient in the p40 chain of IL-12. IL-12 is a heterodimeric molecule, termed IL-12p70, comprising a p40 and a p35 subunit. The p40 chain is shared with another cytokine, IL-23, a heterodimer of IL-12/23 p40 and IL-23 p19. Thus, p40 deficiency also impairs IL-23 signalling. Recent studies have shown that it is indeed IL-23, and not IL-12, that is necessary for the development of EAE [Becher et al. 2002; Gran et al. 2002; Cua et al. 2003]. Also secreted by APCs, IL-23 promotes a unique deviation of Th cytokine secretion, provisionally p 11 termed ThIL-17 (or Th17), characterised by secretion of IL-17, TNF, IL-6, but little IFN- [Iwakura and Ishigame 2006]. The ThIL-17 axis is presently the subject of intense investigation, and its role in the overall Th cell repertoire is still largely undetermined. 2.3. Immunomodulatory treatments for multiple sclerosis Until 1993, there were no disease-modifying treatments for MS; only treatment of acute relapses with methylprednisolone was available. Interferon (IFN)- became the first diseasemodifying treatment for MS, reducing the overall relapse-rate by approximately one-third and probably slowing disease progression [The IFNB Multiple Sclerosis Study Group 1993; Jacobs et al. 1996; PRISMS Study Group 1998]. IFN- treatment also reduces disease activity as evidenced by contrast-enhancing CNS lesions seen on magnetic resonance images (MRI). Since the introduction of IFN- in the treatment of MS, other drugs have been added, such as glatiramer acetate, intravenous immunoglobulins, and mitoxantrone. A common feature of all the disease-modifying MS treatments is that they target the immune system. IFN- is the most widely used first-line drug and will be discussed here in more detail. The exact mechanism of action of IFN- is not known. Research has focused on the immunological steps that have been implicated in MS pathogenesis as outlined in section 2.2. Experimental data indicate that IFN- exerts its action at multiple points of the immune pathogenesis of MS, including (a) T cell activation and co-stimulation by APCs, (b) T cell cytokine secretion, (c) T cell trafficking and CNS transmigration, and (d) effector cell functions within the CNS lesions. These are reviewed in [Zhang et al. 2002] and [Hartung et al. 2004], and will be outlined here: (a) T cell activation and co-stimulation: In order for the myelin-reactive T cells to become activated, APCs must present them with their cognate antigen. Antigen is presented to the T cell receptor in conjunction with the MHC class molecule on the APC. The activation of the T cells depends on co-stimulatory signals from the APC without which the T cells fail to become fully activated or may even become functionally anergic. Although somewhat controversial, most studies have shown that IFN- therapy can counteract some IFN- effects p 12 by reducing co-stimulatory signalling, APC MHC class II molecule expression, IFN-induced MHC class II expression, and APC-mediated IFN- secretion [Yong et al. 1998; Zhang et al. 2002; Hartung et al. 2004]. This, in theory, predicts a reduced capability of the APCs to activate Th cells. (b) T cell cytokine secretion: IFN- therapy has been shown to induce secretion of the immunoregulatory cytokine IL-10 (see section 2.4). Whether IFN- therapy reduces the Th1related IFN- is controversial, and studies have found both increased and decreased secretion of Th1 cytokines during IFN- therapy [Dayal et al. 1995; Rudick et al. 1998; Gayo et al. 1999; Khademi et al. 2000; Wandinger et al. 2001; Mirowska et al. 2003]. Some have found increased secretion of the Th2 cytokine IL-4, but others have found no effects [Khademi et al. 2000;Mirowska et al. 2003;Rudick et al. 1998]. In summary, although a Th2-promoting effect of IFN- is often postulated, previous studies have not clearly verified this. (c) T cell trafficking and CNS transmigration: Lymphocyte trafficking is controlled by the expression of adhesion molecules, chemokines, chemokine receptors, and the activation stage of the cell. While IFN- therapy reduces the numbers of systemically circulating lymphocytes, the specific effects are controversial. Entry into the CNS requires the sequential action of selectins, integrins, chemokines, and matrix metalloproteinases (MMPs), enabling the lymphocytes to migrate across the blood-brain barrier (BBB)[Ransohoff et al. 2003; Sellebjerg and Sorensen 2003]. Studies have shown decreased, unchanged, or even increased expression of the CNS-homing chemokine receptors CXCR3 and CCR5 upon IFN therapy (discussed in P-II). The CCR5 ligands, CCL3 and CCL5 have been found to decrease upon IFN- therapy [Zang et al. 2001]. IFN- has been shown to decrease expression of some of the molecules involved in CNS transmigration, VLA-4 [Calabresi et al. 1997a; Chabot et al. 1997; Muraro et al. 2000] and MMPs [Leppert et al. 1996; Stuve et al. 1996; Bartholome et al. 2001]. Conversely, IFN- increases levels of the soluble VCAM molecule, blocking interactions between lymphocyte VLA-4 and endothelial VCAM, consequently decreasing lymphocyte entry into the CNS [Calabresi et al. 1997b; Kallmann et al. 2000; Muraro et al. 2000; Jensen et al. 2005]. p 13 (d) Effector cell functions within the CNS lesions: For ethical reasons, CNS biopsies in the study of MS are obviously not readily available and few studies have addressed the effects of IFN- in the CSF and the inflammatory MS lesions. It is unlikely that IFN- can enter the CNS through an intact BBB in therapeutically relevant quantities. While MS is characterised by a focally compromised BBB, it is theoretically possible that IFN- can diffuse through the BBB at these points. However, as IFN- has a BBB-stabilising effect, effectively restoring the BBB, this route of entry of IFN- into the CNS is probably blocked shortly after treatment initiation [Kraus et al. 2004]. As outlined above, IFN- apparently acts in an unspecific manner on many components of the immune system, making it difficult to pinpoint the effects that are most crucial to the therapeutic efficacy. Apart from the above-mentioned effects, IFN- also affects trafficking and activation of immune cells other than mononuclear cells. Accordingly, granulocytes are affected, as is the hypothalamic-pituitary-adrenal hormonal axis [Goebel et al. 2005]. The therapeutic implications of these effects are unknown. 2.4. Biomarkers New and emerging MS treatments have provided the clinician with a palette of therapeutic options, and these will probably increase dramatically in the near future. It is therefore increasingly important to be able to tailor the pharmacological treatment to the needs of the individual patient. As MS disease progression is slow, abrupt, and sometimes obscured by the plasticity of the CNS, clinical assessment of disease activity is not feasible as a marker of inflammatory activity, thus creating the need for surrogate biomarkers. Biomarkers are defined as “A characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention” [Biomarkers Definitions Working Group 2001]. Biomarkers can be divided into type 0 biomarkers that are measures of the natural history of a disease, e.g. indicators of disease activity or prognosis, and type 1 biomarkers that indicate an intervention effect, e.g. a drug [Bielekova and Martin 2004]. By p 14 definition, type 1 biomarkers are not required to reflect biologically relevant effects, as long as the biomarker is influenced by the treatment in question. Although never formally defined, type 1 biomarkers might be sub-divided into biomarkers that do reflect treatment effects that are pathophysiologically relevant to the disease in question, and biomarkers that do not, i.e. markers that do not correlate with clinical outcome in the natural history of the disease; the latter merely measuring a biological response to the treatment. 2.4.1. Type 0 biomarkers in multiple sclerosis Studies have shown aberrant low-grade immune activation in MS, both systemically and intrathecally. Several studies have addressed the possibility of using measures of immune activation as a predictor of MS disease severity, prognosis, or activity, i.e. a type 0 biomarker. Indeed, correlations with disease activity and severity have been found among immunological markers related to T cell effector functions, activation, trafficking, and CNS transmigratory potential. Some of those most frequently mentioned are outlined below and summarised in table 1: CCR5 The chemokine receptor CCR5 binds the chemokines CCL3, CCL4, and CCL5, and is expressed primarily by Th1 cells [Bonecchi et al. 1998;Loetscher et al. 1998]. It is markedly over-expressed on T cells in MS CNS lesions [Balashov et al. 1999; Sorensen et al. 1999], and MS patients have higher numbers of CSF CD4+ T cells expressing CCR5 at relapse than at remission [Misu et al. 2001]. Hetero- and homozygosity in MS patients for a genetic polymorphism that encodes a non-functional truncated CCR5 protein (CCR532) has been reported to result in a milder clinical course and delayed onset [Barcellos et al. 2000; Sellebjerg et al. 2000; Kantor et al. 2003], but this has been challenged in later studies [Silversides et al. 2004; Kantarci et al. 2005]. CXCR3 Another chemokine receptor, CXCR3, is also linked to Th1 effector functions [Bonecchi et al. 1998], and 99 % of T cells in the perivascular cuff of active MS lesions express CXCR3 p 15 [Sorensen et al. 1999]. T cell CXCR3 expression has also been shown to correlate with disease activity [Misu et al. 2001; Sindern et al. 2002; Mahad et al. 2003; Nakajima et al. 2004]. VLA-4 T cell expression of the integrin VLA-4 is important for leukocyte tethering to CNS endothelial vascular cell adhesion molecule (VCAM) and subsequent transendothelial migration into the CNS. Increased expression of Th cell VLA-4 has been linked with disease activity [Barrau et al. 2000]. Substantiating the role of VLA-4 in MS pathogenesis, a recently approved RRMS treatment, an antibody blocking the interaction between T cell VLA-4 and endothelial VCAM, has been shown to markedly reduce the relapse-rate in MS patients [Polman et al. 2006]. Interleukin-10 IL-10 is a regulatory cytokine with multiple effects on APC and lymphocyte functions, including inhibition of antigen-specific T cell-proliferation, co-stimulation, and cytokine secretion [Taylor et al. 2006]. PBMC synthesis of IL-10 is negatively correlated with MS disease activity, as demonstrated by in vivo and in vitro assays. IL-10 levels are lower in active MS and prior to relapses than at remission [Rieckmann et al. 1994; Musette et al. 1996; Boxel-Dezaire et al. 1999], and constitutively low in SPMS patients [Correale et al. 1995; Rieckmann et al. 1995; Boxel-Dezaire et al. 1999]. Interleukin-12 As described in section 2.2., the immune response in MS has long been considered to be Th1-deviated. Since IL-12 is a powerful inducer of Th1 it has been studied as a possible biomarker in MS. Interestingly, to my knowledge only one study has shown correlations between monocyte IL-12 production and disease activity measures [Makhlouf et al. 2001]. In the light of the newly discovered IL-23/ThIL17 axis, which appears to be more important than the IL-12/Th1 axis in autoimmune diseases, this is perhaps not surprising. p 16 Matrix metalloproteinase 9 and Tissue inhibitor of metalloproteinase 1. The balance of the levels of MMP-9 and TIMP-1 in serum and CSF has also been scrutinised as a potential type 0 biomarker in MS. MMP-9 (Gelatinase B) is involved in breakdown of the blood-brain barrier and lymphocyte migration in the CNS parenchyma whereas TIMP-1 acts as an inhibitor of the matrix metalloproteinases such as MMP-9. Although some have found correlations between MS disease activity measures and MMP-9 / TIMP-1 levels, research in this field has been hampered by methodological problems due to the fact that serum samples are easily contaminated with MMP-9 derived from ex vivo white blood celllysis and thrombocyte activation [Gerlach and Tanus-Santos 2006]. 2.4.2. Type 1 biomarkers in multiple sclerosis While the above-mentioned immunological parameters are putative type 0 biomarkers, efforts have also been made to assess the efficacy of IFN- treatment by the measurement of immunological surrogate biomarkers, i.e. type 1 biomarkers. Several markers have been investigated, ranging from concentrations of plasma chemokines, cytokines, or other proteins to measurements of messenger RNA (mRNA) encoding these proteins in leukocytes. Some of the proposed type 1 biomarkers have also been suggested as type 0 biomarkers, thus linking the properties of a type 1 biomarker with the underlying disease pathophysiology. As for IFN- type 1 biomarkers with no direct relation to MS pathophysiology, these include the so-called interferon-stimulated genes (MxA protein, 2-microglobulin, 2'5'oligoadenylate synthetase (OAS), etc). They are useful when assessing the bioavailability of INF- in MS patients where the formation of circulating neutralising antibodies may prevent binding of IFN- to its receptors and, consequently, abolish the pharmacological effect of IFN-. However, these markers carry no information regarding the prognosis or clinical response to the treatment, and they will not be discussed further here. IFN- type 1 biomarkers that may affect the underlying, pathophysiologically relevant processes are numerous, and include IL-10, CXCL10 (IP-10), and Tumour necrosis factor-Related Apoptosis Inducing Ligand (TRAIL). These molecules are all induced by p 17 IFN- treatment. Due to the immune-regulatory properties of IL-10, it is a particularly attractive candidate as a type 1 biomarker with relation to MS pathophysiology since it is also induced by IFN- treatment [Rudick et al. 1998; Rep et al. 1999; Liu et al. 2001]. CXCL10, a chemokine with specificity for the Th1-associated chemokine receptor CXCR3 is transiently increased by IFN- therapy [Buttmann et al. 2004]. As CXCL10 is also elevated in the CSF of MS patients, and has been implicated in the chemo-attraction of T cells to the CNS, increased plasma concentrations of CXCL10 could result in disruption of the biologically active concentration gradient, or in the downregulation of CXCR3 on the T cell surface [Trebst and Ransohoff 2001]. TRAIL, a member of the TNF superfamily, has apoptosis-inducing and immunoregulatory properties [Wiley et al. 1995; Song et al. 2000; Hilliard et al. 2001; Lunemann et al. 2002], and has also been suggested as a type 1 IFN- response biomarker in MS [Wandinger et al. 2003; Gilli et al. 2006]. However, the relationship between TRAIL expression and disease activity in untreated MS has not been studied. Suggested molecules Type 0 biomarkers CCR5, CXCR3, VLA-4, IL-10, IL-12, MMP9/TIMP-1 Type 1 biomarkers with no relation to MS MxA, 2-microglobulin, OAS pathophysiology Type 1 biomarkers with relation to MS IL-10, CXCL10 (IP-10); TRAIL pathophysiology Table 1: Commonly suggested biomarkers in MS and IFN- therapy. p 18 3. Aim of the thesis The clinical management of MS patients is challenging due to the inherent inter-patient heterogeneity of disease presentation, clinical course, prognosis, and responsiveness to therapy. Additionally, the long-term effects of fluctuations in biological disease activity or treatment efficacy are manifested clinically with a delay of up to several years, consequently narrowing the clinician’s window of opportunity to intervene with a rational therapeutic strategy at the appropriate time. The overall aim of my studies was to identify possible surrogate markers of disease activity (type 0 markers) and treatment efficacy (type 1 markers), thus providing the initial steps towards future paraclinical tools to aid in the individualised management of patients with multiple sclerosis. A secondary aim of the studies was to shed more light on the basic immunopathology of multiple sclerosis by studying in vivo lymphocyte activation in untreated and IFN--treated patients. P-I was designed to identify and characterise a subset of CD4+ T helper cells that is biologically relevant in MS pathogenesis. To provide information regarding T cell trafficking during IFN- therapy P-II studied chemokine and chemokine receptor expression from the level of gene expression to T cell surface protein expression, and finally soluble protein levels in MS patients before and after treatment with IFN-. In order to clarify the effects of IFN- therapy on Th1 / Th2 immune deviation P-III studied in vivo PBMC gene expression of cytokines and transcription factors relevant to the Th1 / Th2 dichotomy in MS patients and assessed changes induced by IFN- therapy. p 19 4. Summary of own studies 4.1. P-I – CD26 paper Materials and methods: Peripheral blood CD4+ T cells from healthy controls and RRMS patients were analysed by flow cytometry. Expression of a panel of surface markers of memory / naïve status, activation, Th1 / Th2 functions, co-stimulation, migration, and apoptosis was studied in CD4+ T cell subsets defined by their expression of the memory cell marker CD45R0 and the activation marker CD26. Plasma concentrations of the apoptosisrelated proteins Fas and Fas-Ligand were measured by ELISA. MS patients were re-studied after three months of IFN- therapy. Results: When dividing CD4+ T cells according to their expression of CD45R0 and CD26 four major subsets were identified: CD45R0-CD26intermediate cells, CD45R0+CD26low cells, CD45R0+CD26intermediate cells, and CD45R0+CD26high cells. The latter subset was enriched for cells expressing markers of T effector memory cells, activation, Th1 effector functions, and cells with a chemokine receptor expression pattern favouring CNS homing. MS patients expressed lower levels of CCR4 (a Th2-related chemokine receptor), PD-1 (a co-inhibitory signalling molecule), and L-selectin (favouring homing to lymph-nodes) in the CD4+ CD45R0+ CD26high subset and had lower levels of soluble Fas-Ligand compared with healthy controls. The numbers of circulating CD4+ CD45R0+ CD26high cells correlated with clinical MS disease severity. In the same cell-subset, IFN- therapy caused increased expression of the chemokine receptors CCR4, CCR5, CCR7, and the apoptosis-related Fasmolecule. Plasma levels of soluble Fas and Fas-Ligand also increased. 4.2. P-II – Chemokine paper Materials and methods: Blood samples from RRMS patients and healthy controls were analysed for chemokine and chemokine receptor expression. MS patients were re-sampled after three months of IFN- therapy. To address the biological effects of the pharmacokinetics of IFN- treatment an additional cohort of MS patients was included in p 20 which the timing of blood sampling in relation the latest injection of IFN- was standardised (9-12 hours post-injection). Gene-expression of the chemokine receptors CCR4, CCR5, CCR7, and CXCR3 in purified PBMCs was quantified along with markers of Th1, Th2, and regulatory functions. Flow cytometry was used to quantify CD4+ T cell chemokine receptor surface expression, and concentrations of soluble plasma chemokines (CCL3, CCL4, CCL5, CXCL9, and CXCL10) were measured in ELISA assays or a cytometric bead array. Results: MS patients and healthy controls showed comparable levels of soluble chemokines, chemokine receptor gene expression, and CD4+ surface chemokine receptor expression except for CD4+ surface expression of CCR4, which was lower in MS patients. IFN- treatment increased the CD4+ surface expression of CCR4, CCR5, and CCR7 at steady state conditions. In contrast, when samples were timed at 9-12 hours post-IFN--injection CCR4, CCR5, and CCR7 surface expression was unaltered while CXCR3 expression was significantly decreased. There were no fluctuations in chemokine receptor gene-expression or soluble chemokine concentrations during IFN- therapy except for CXCL10 where both mRNA and plasma protein concentrations were increased and tightly correlated. CCR4 and CCR7 mRNA levels correlated with mRNAs of markers of Th2 and regulatory functions while CCR5 mRNA correlated with Th1 markers. 4.3. P-III – Cytokine paper Materials and methods: Gene expression in PBMCs from RRMS patients (before and after three months of IFN- therapy) and healthy controls was analysed using real-time reverse transcriptase PCR. Genes tested included cytokines belonging to Th1 or Th2 functions and related transcription factors. A newly discovered axis of Th cell immune activation, termed ThIL-17, was also addressed by measuring gene expression of IL-23 and IL-17. mRNA for the regulatory genes, IL-10 and TGF-, was quantified as well. Results: IL-23 mRNA was increased in MS patients compared to healthy controls. IFN- therapy resulted in increased IL-10 mRNA expression and slightly decreased IL-13 mRNA expression. IL-23 mRNA expression remained elevated during IFN- therapy. None of the p 21 classical Th1 cytokines were affected by IFN- treatment nor did we find any significant change in mRNA levels of transcription factors involved in the Th1 / Th2 immune deviation. We found a strong correlation between mRNAs for putative Th1 markers and the classical Th1 cytokine IFN- and between mRNAs for putative Th2 markers and the classical Th2 cytokine IL-4. IL-10 mRNA expression was completely independent of the expression of any Th1- or Th2-related cytokines. 4.4. Overall conclusions of own studies The studies suggested the existence of a CD4+ T cell subset that is enriched for many of the surface markers associated with proinflammatory responses and that this subset correlates with clinical MS disease measures. Elevated levels of IL-23 mRNA was found in PMBC’s from untreated MS patients, who also had decreased CD4+ T cell surface expression of the Th2-related chemokine receptor CCR4. IFN- treatment caused acute induction of IL-10 and CXCL10 mRNA in PBMCs and decreased CD4+ T cell surface expression of the Th1related chemokine receptor CXCR3. More long-term “steady state” effects included increased CD4+ T cell surface expression of the chemokine receptors CCR4, CCR5, and CCR7. We also observed complex effects on the expression of the apoptosis-related molecules fas and fas-ligand during IFN- therapy. Treatment with IFN- did not normalise the increased IL-23 PBMC mRNA expression. p 22 5. Critique of materials and methods 5.1. General methodological considerations Due to the tremendous complexity of mammalian immunology, it is often necessary to focus on a small part of the immune system when conducting a scientific study. When doing this, it is inherently difficult to take into account the effects of the multitude of communication pathways between the different players in the immune system. Consequently, conclusions drawn from studies of isolated immune mechanisms should always be made with caution. This is readily demonstrated by previous immunological studies in MS, where results are often seemingly contradictive and difficult to reproduce. Not surprisingly, even subtle differences in the experimental design can sometimes affect the findings and conclusions of the studies. It must be stressed that the study of the mechanism of action of IFN- and other immunomodulatory treatments is hampered by several pitfalls. Firstly, many studies are based on in vitro assays or the MS animal model, EAE. Experience shows that it is not always possible to extrapolate results from in vitro or animal conditions to humans. Secondly, even in vivo studies in humans sometimes produce inconsistent results probably due to methodological differences, and may also report biological treatment effects that are unrelated to the therapeutic efficacy of the drug (as discussed in section 2.4). In an attempt to address the immunology in MS in the most direct manner, our studies have focused on human in vivo conditions rather than animal studies or in vitro assays. This approach creates its own set of challenges. The systemic inflammatory response in MS is long-term and low-grade, unlike such conditions as septicaemia or bacterial meningitis that involve massive inflammation. Hence, systemic immunological changes in MS are often subtle and therefore easily overlooked. A much more clear picture can often be obtained using in vitro assays where experimental conditions can be designed to yield a much more robust and conclusive outcome. It must therefore be stressed that animal and in vitro studies are also highly warranted since they provide other invaluable insights in p 23 immunology. The challenge is, however, to extrapolate such experimental data to in vivo human conditions, and conclusions must be made from a synthesis of animal studies, in vitro, and in vivo findings. In our studies, we have focused attention on CD4+ T (Th) cells as they are often thought to be critically involved in MS pathogenesis. However, it is highly unlikely that MS is purely the result of aberrant Th cell behaviour. Although Th cells are probably key players in MS pathogenesis, they constitute only part of the picture. The conclusions to the findings on Th cell biology in MS must therefore be viewed in the context of the complexity of the immune system. An important finding in P-II and P-III was that the timing of blood sampling in relation to the patients’ most recent IFN- injection significantly affected the measurements of gene expression, surface protein expression, and soluble plasma protein concentrations. While this offers a possible explanation for some of the discrepancies in the previously reported immunological effects of IFN-, it is also a potential methodological problem in studies P-I and P-III. These studies did not include a cohort of patients in which blood samples were obtained at a standardised time after IFN- injection, and may consequently have missed some of the acute IFN--induced effects. Taken together, this underlines that the timing of sample acquisition is a crucial consideration in future studies concerning the immunological effects of any pharmacological treatment. 5.2. Technical considerations Flow cytometry is a powerful immunological tool by which the characteristics of single cells can be analysed in a number of different ways, including surface and intracellular molecule expression, cell-cycle stages, proliferation history, apoptosis and viability, calcium-flux, etc. Many of these measures are obtained by cell staining with fluorochrome-conjugated antibodies. Significant inter-study variability in the quantitative measurements of e.g. surface molecule expression can be expected due to the use of different equipment, antibody preparations, antibody conjugations, and antibody concentrations. Variability can also arise p 24 from the use of different procedures when handling cells prior to and during analysis: sampling conditions, temperature conditions, staining protocols, etc. Hence, it is not always possible to translate flow cytometric results from one study to another. In the flow cytometry analyses that form the basis of this thesis, intra-assay variability was minimised by calibrating the flow cytometer before each run, titrating antibodies prior to use, normalising expression to that of isotype-matched control antibodies, and by comparing results between patient groups (healthy controls, untreated MS patients, IFN--treated MS patients) analysed with the same equipment and reagents. Real-time quantitative reverse transcriptase PCR is a relatively new modality of PCR that has eased the quantification of gene expression compared to earlier semiquantitative gel-based methods that are rather complicated and laborious. Although a simpler procedure, quantitative RT-PCR still involves many steps and, consequently, many inherent sources of variability of the results: RNA stabilisation and storage, RNA purification, reverse transcription of RNA into cDNA, differential PCR efficiency and the choice of “housekeeping-gene” i.e. a constitutively expressed endogenous control gene. All these factors contribute to the overall analytical variability, and, consequently, decrease the statistical power to detect minor differences between samples. Theoretically, this does not increase the risk of false positive findings but does increase the risk of false negative results. Consequently, some relevant biological effects of IFN- therapy may have been overlooked in our studies. However, it could be argued that biologically relevant changes in gene expression ought to be of a certain magnitude that renders them relatively robust to analytical inaccuracies. In P-III we addressed cytokine and transcription factor mRNA levels indicative of Th1 or Th2 patterns. In support of the validity of the data is the fact that we found very close correlations between cytokines and transcription factors related to Th1 and Th2, respectively. However, in order to get more detailed information, it would be preferable to study not only mRNA expression in a bulk preparation of PBMCs but also in separated mononuclear cell subsets such as CD4+ T cells, CD8+ T cells, monocytes, NK cells, and B cells. Moreover, some cytokine mRNAs could not be detected in the PCR assay, underlining the inherent difficulties in measuring immune effector functions in a setting of low-grade inflammation, p 25 such as MS. Ideally, the in vivo quantification of mRNAs for cytokines should be supplemented with in vitro stimulation assays with anti-CD3 and anti-CD28 antibodies, putative MS autoantigens, and control antigens. p 26 6. Discussion 6.1. Immunological characteristics of untreated MS patients Aberrant immune activation is an established finding in MS. There is, though, some debate as to whether the immune activation is part of the aetiology of MS or if it merely reflects an epiphenomenon in response to an underlying degenerative disease in the CNS. However, if MS pathogenesis resembles that of the animal model EAE it is likely that the aberrant immune activation in MS is indeed pathogenic. As previously discussed, the HLA-DR2 linkage substantiates this notion. The fact that all clinically efficacious therapies act by targeting the immune system also points to a central role of the immune system in MS pathophysiology. In study P-I, the identification of an activated Th memory cell subset (CD4+ CD45R0+ CD26high) that correlates with clinical disease severity is indicative of a close relation between systemic immune activation and clinical disease. Correlations between CD26 expression and markers of disease activity have previously been reported [Constantinescu et al. 1995; Khoury et al. 2000; Jensen et al. 2004]. Further substantiating the link between clinical disease and the CD4+ CD45R0+ CD26high cells, other immunological markers that have previously been correlated with disease activity and/or severity were all enriched in this Th cell subset. These markers are all functionally linked to a proinflammatory immune response and a CNS transmigratory phenotype of the lymphocytes. Underlining the pathogenic potential of this subset, some of the markers, CCR5, CXCR3, and CXCR6, have been specifically linked to Th1 effector functions [Bonecchi et al. 1998; Loetscher et al. 1998; Calabresi et al. 2002]. Future studies should re-assess these markers in the light of the newly discovered IL-23/ThIL-17 axis. In MS patients, cells in the CD4+ CD45R0+ CD26high subset expressed less L-selectin, PD-1, and CCR4 compared with healthy controls. This is consistent with a population of cells that is more activated, less prone to inhibitory signalling, and with reduced potential for a presumably less detrimental Th2 response, respectively. While the decreased expression of L-selectin and PD-1 in MS patients has not previously been reported, decreased expression of CCR4 on total CD4+ T cells has been reported [Misu et al. 2001; Matsui et al. 2004]. This indicates a p 27 proinflammatory immune deviation with preponderance for CNS homing in MS patients. These findings give reason for the development of immunomodulatory treatments that attempt to dampen inflammatory responses and CNS homing mechanisms. In P-III, increased expression of IL-23 mRNA was found in MS patients. As described in section 2.2., IL-23 belongs to a newly defined proinflammatory axis, termed ThIL-17 that differs somewhat from the classical Th1 axis and has been found to be crucial in EAE pathogenesis. Recently, another study has confirmed our finding that IL-23 is increased in MS [Vaknin-Dembinsky et al. 2006]. In that study, monocyte-derived dendritic cells from MS patients secreted increased amounts of IL-23, and expressed increased IL-23 p19 mRNA. Correspondingly, the authors also found increased T cell expression of IL-17, a molecule we were unable to detect in the gene-expression analyses used in P-III. The findings in P-III and the recent work by Dembinsky et al support the proposed crucial role of IL-23 in MS and, possibly, other autoimmune conditions, and challenge the currently accepted role of the Th1 axis in MS immunopathogenesis. However, much further research into the IL-23 / ThIL-17 axis is needed in order to establish its role in inflammatory conditions in health and disease. 6.2. Immunological effects of IFN- therapy in MS As outlined in section 2.3., the effects of IFN- therapy are complex, and the study of these effects is hampered by several pitfalls. P-I, P-II and P-III addressed some of the previously proposed effects of IFN- therapy. P-II established that CD4+ T cell surface chemokine receptor expression is modulated by IFN- therapy. Expression of the Th1-associated chemokine receptor CCR5 was increased by IFN-, but this was also true for the Th2-related CCR4. In addition, CCR7, a lymph-node homing receptor, was also increased by IFN- treatment. CD4+ T cell surface expression of CXCR3 was unaltered by IFN- in the initial data analysis. Previous studies have addressed IFN--induced effects on the expression of some of the chemokine receptors that were studied in P-III. The findings have been controversial. p 28 One study found no effects on CD4+ T cell expression of CXCR3, while another found decreased surface expression during IFN- therapy [Kivisakk et al. 2003; Sorensen and Sellebjerg 2002]. The same discrepancies apply to CCR5 expression, which has been found to increase, decrease, or remain unaltered during IFN- therapy [Wandinger et al. 2001; Zang et al. 2001; Sorensen and Sellebjerg 2002; Kivisakk et al. 2003]. To my knowledge, interferon--induced effects on CD4+ T cell expression of CCR4 and CCR7 have previously not been studied. In the initial analysis of the data in P-II, we found no effects of IFN- therapy on CD4+ T cell CXCR3 expression, and an increased expression of CCR4, CCR5, and CCR7. However, we noticed that there are considerable effects of the timing of blood sampling in relation to an IFN- injection. In contrast to the “steady state” findings mentioned above, a cohort of patients sampled shortly after an IFN- injection (9-12 hours) did indeed show suppression of CXCR3 surface expression while expression of CCR4, CCR5, and CCR7 was unaltered. These findings demonstrate that IFN- treatment induces a complex shift in the expression of different chemokine receptors, which fluctuates according to the pharmacokinetics of an IFN- injection. It was beyond the scope of the descriptive study design in P-II to investigate the functional significance of the fluctuations of CCR4, CCR5, CCR7, and CXCR3 expression. However, the data do indicate that IFN- therapy induces changes in lymphocyte trafficking, which might affect CNS T cell migration and, consequently, the inflammatory environment in the CNS lesions. P-II also provides at least a partial explanation for the controversial findings in the previous literature, namely the importance of the timing of the blood sampling versus the latest IFN- injection. In P-I, IFN- therapy caused increased surface expression of CCR4, CCR5, CCR7, and the apoptosis-inducing Fas in the putative pathogenic Th cell subset (CD4+ CD45R0+ CD26high). The functional implications of these findings are difficult to predict, but IFN- therapy seems to have broad effects on lymphocyte trafficking and apoptosis pathways, particularly in a Th subset that is enriched for markers of many effector functions implicated in MS pathophysiology. Functional characterisation of the CD4+ CD45R0+ CD26high cells is warranted to further clarify their role in MS. p 29 P-III examined changes in gene expression of cytokines and transcription factors relating to Th1, Th2, or regulatory functions induced by IFN- therapy. Patients were studied 1-7 days after their latest IFN- injection. In this study, apart from marked increases in IL-10 mRNA (regulatory cytokine) only marginal fluctuations in Th1- or Th2-related markers were observed during IFN- therapy. The IFN--induced IL-10 expression has been found by several others while the suggested Th2-deviating effects of treatment are far more controversial. We could not confirm a Th2-deviating effect of IFN- therapy. Although some previous studies on the Th1 / Th2 modulating properties of IFN- treatment have largely reported Th2-deviating effects (see section 2.3.), this claim has been challenged by other studies [Khademi et al. 2000; Wandinger et al. 2001]. P-III adds to the view that IFN- therapy is not merely skewing the immune system from a Th1-biased response towards a Th2 pattern, and indicates that the effects are more complex. The ThIL-17-related cytokine IL-23 mRNA, which was found to be elevated in untreated MS patients, was not modulated by IFN- therapy. As the ThIL-17 axis is putatively important in MS pathogenesis, it is interesting that IFN- therapy does not appear to affect it. It is possible that future MS therapies will attempt to target the IL-23/ThIL-17 axis. However, much work needs to be done to establish the role of the ThIL-17 system in MS and other immune-medicated diseases before devising such treatments [Bowman et al. 2006]. A key question is whether the clinical benefit of IFN- therapy is mediated through the acute effects that are in play during the first hours after the injection or the subtler “steady state” effects. An indication of this may come from the study of correlations between type 0 and type 1 biomarkers. The biomarkers that share type 0 and type 1 properties can be evaluated for “acute” or “steady state” modulation, thereby providing a clue to whether acute, or steady state effects are more important. IL-10 is a biomarker with both type 0 and type 1 properties, and as we demonstrated in P-III, it is induced only during the first 1-2 days following an IFN- injection. A similar pattern is observed regarding the chemokine CXCL10 (P-II), which has also been linked to MS pathophysiology [Sorensen 2004]. This provides an indication that the acute IFN- effects, not the steady state effects, are most likely to be the therapeutically relevant for the clinical response in MS patients. Future study p 30 designs should consider such issues concerning the kinetics of the pharmacological effects induced by an IFN- injection. 6.3. Putative biomarkers in MS and IFN- therapy As discussed earlier, the best type 1 markers for use in the clinical management of MS are theoretically those that act as not only type 1 biomarkers but are also biologically linked to MS pathophysiology. Among numerous putative biomarkers reported in the literature, findings regarding IL-10 and CXCL10 have been most consistent and uncontroversial, and both may have strong impacts on central mechanisms in the immune response involved in MS. P-II and P-III have substantiated this finding, and prospective studies on these markers as type 1 markers in IFN- therapy are warranted. P-I points to the CD4+ CD45R0+ CD26high subset as a putative type 0 biomarker, and this subset could also be investigated for other type 0 markers, e.g. markers of activation, apoptosis, effector functions, and migration. Looking for markers that reflect Th2-deviation induced by IFN- therapy may not be feasible as the biological effects of IFN- are not strictly Th2-promoting, as demonstrated in P-III. Alternatively, the newly discovered IL-23/ThIL-17 axis should be evaluated for useful type 0 and type 1 biomarkers, as this axis is a promising candidate as a key player in autoimmune conditions such as MS. In conclusion, larger prospective studies that also include some of the presently available biomarkers such as new gadolinium-enhancing MRI lesions are needed. We have already launched such studies. Parallel to clinical validation of biomarkers it is also relevant to apply functional testing of some of the candidate markers in order to discriminate epiphenomena from immunological markers of biological relevance to MS pathophysiology. p 31 7. Future perspectives I believe that future pharmacological MS therapy will involve tailoring to the individual needs of the patients and the use of combinations of immunomodulatory treatments; possibly supplemented by neuroprotective drugs and drugs that promote CNS repair processes. Understanding the immune pathophysiology behind MS as well as monitoring disease activity and prognostic stratification will be crucial instruments in this process, and will provide a basis for earlier and more efficient MS therapy than hitherto available. It will be interesting to follow the development of MS therapeutics and tools for disease monitoring, as I believe both will take a leap forward in the course of the next decade. p 32 8. References Amyes E, McMichael AJ, Callan MFC. Human CD4(+) T cells are predominantly distributed among six phenotypically and functionally distinct subsets. Journal of Immunology 2005; 175: 5765-5773. Appay V. The physiological role of cytotoxic CD4+T-cells: the holy grail? Clinical and Experimental Immunology 2004; 138: 10-13. Balashov KE, Rottman JB, Weiner HL, Hancock WW. CCR5(+) and CXCR3(+) T cells are increased in multiple sclerosis and their ligands MIP-1alpha and IP-10 are expressed in demyelinating brain lesions. Proc.Natl.Acad.Sci.U.S.A 1999; 96: 6873-6878. Banerjee D, Liou HC, Sen R. c-ReI-dependent priming of naive T cells by inflammatory cytokines. Immunity 2005; 23: 445-458. Barcellos LF, Schito AM, Rimmler JB et al. CC-chemokine receptor 5 polymorphism and age of onset in familial multiple sclerosis. Multiple Sclerosis Genetics Group. Immunogenetics 2000; 51: 281-288. Barrau MA, Montalban X, Saez-Torres I, Brieva L, Barbera N, Martinez-Caceres EM. CD4(+)CD45RO(+)CD49d(high) cells are involved in the pathogenesis of relapsing-remitting multiple sclerosis. J.Neuroimmunol. 2000; 111: 215-223. Bartholome EJ, Van A, I, Koyen E et al. Human monocyte-derived dendritic cells produce bioactive gelatinase B: inhibition by IFN-beta. J.Interferon Cytokine Res. 2001; 21: 495-501. Becher B, Durell BG, Noelle RJ. Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J.Clin.Invest 2002; 110: 493-497. Bielekova B, Martin R. Development of biomarkers in multiple sclerosis. Brain 2004; 127: 1463-1478. p 33 Bielekova B, Sung MH, Kadom N, Simon R, McFarland H, Martin R. Expansion and functional relevance of high-avidity myelin-specific CD4(+) T cells in multiple sclerosis. Journal of Immunology 2004; 172: 3893-3904. Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin.Pharmacol.Ther. 2001; 69: 89-95. Bonecchi R, Bianchi G, Bordignon PP et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J.Exp.Med. 1998; 187: 129-134. Bowman EP, Chackerian AA, Cua DJ. Rationale and safety of anti-interleukin-23 and antiinterleukin-17A therapy. Curr.Opin.Infect.Dis. 2006; 19: 245-252. Boxel-Dezaire AH, Hoff SC, van Oosten BW et al. Decreased interleukin-10 and increased interleukin-12p40 mRNA are associated with disease activity and characterize different disease stages in multiple sclerosis. Ann.Neurol. 1999; 45: 695-703. Brønnum-Hansen H, Koch-Henriksen N, Stenager E. Trends in survival and cause of death in Danish patients with multiple sclerosis. Brain 2004; 127: 844-850. Burns J, Bartholomew B, Lobo S. Isolation of myelin basic protein-specific T cells predominantly from the memory T-cell compartment in multiple sclerosis. Annals of Neurology 1999; 45: 33-39. Buttmann M, Merzyn C, Rieckmann P. Interferon-beta induces transient systemic IP10/CXCL10 chemokine release in patients with multiple sclerosis. J.Neuroimmunol. 2004; 156: 195-203. Calabresi PA, Pelfrey CM, Tranquill LR, Maloni H, McFarland HF. VLA-4 expression on peripheral blood lymphocytes is downregulated after treatment of multiple sclerosis with interferon beta. Neurology 1997a; 49: 1111-1116. Calabresi PA, Tranquill LR, Dambrosia JM et al. Increases in soluble VCAM-1 correlate with a decrease in MRI lesions in multiple sclerosis treated with interferon beta-1b. Ann.Neurol. 1997b; 41: 669-674. p 34 Calabresi PA, Yun SH, Allie R, Whartenby KA. Chemokine receptor expression on MBPreactive T cells: CXCR6 is a marker of IFNgamma-producing effector cells. J.Neuroimmunol. 2002; 127: 96-105. Chabot S, Williams G, Yong VW. Microglial production of TNF-alpha is induced by activated T lymphocytes. Involvement of VLA-4 and inhibition by interferonbeta-1b. J.Clin.Invest 1997; 100: 604-612. Confavreux C, Compston A. The Natural History of Multiple Sclerosis. In: Compston A, McDonald I, Noseworthy J et al, editors. McAlpine's Multiple Sclerosis. Churchill Livingstone, 2005: 183-272. Confavreux C, Vukusic S. Natural history of multiple sclerosis: a unifying concept. Brain 2006; 129: 606-616. Constantinescu CS, Kamoun M, Dotti M, Farber RE, Galetta SL, Rostami A. A longitudinal study of the T cell activation marker CD26 in chronic progressive multiple sclerosis. J.Neurol.Sci. 1995; 130: 178-182. Cooke SF, Bliss TVP. Plasticity in the human central nervous system. Brain 2006; 129: 16591673. Correale J, Gilmore W, Mcmillan M et al. Patterns of Cytokine Secretion by Autoreactive Proteolipid Protein-Specific T-Cell Clones During the Course of Multiple-Sclerosis. Journal of Immunology 1995; 154: 2959-2968. Cua DJ, Sherlock J, Chen Y et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003; 421: 744-748. Dayal AS, Jensen MA, Lledo A, Arnason BG. Interferon-gamma-secreting cells in multiple sclerosis patients treated with interferon beta-1b. Neurology 1995; 45: 2173-2177. Delprete G, Decarli M, Almerigogna F, Giudizi MG, Biagiotti R, Romagnani S. Human Il-10 Is Produced by Both Type-1 Helper (Th1) and Type-2 Helper (Th2) T-Cell Clones and Inhibits Their Antigen-Specific Proliferation and Cytokine Production. Journal of Immunology 1993; 150: 353-360. p 35 Ebers GC, Sadovnick AD. The Geographic-Distribution of Multiple-Sclerosis - A Review. Neuroepidemiology 1993; 12: 1-5. Ford ML, Evavold BD. Specificity, magnitude, and kinetics of MOG-specific CD8(+) T cell responses during experimental autoimmune encephalomyelitis. European Journal of Immunology 2005; 35: 76-85. Fujinami RS, Von Herrath MG, Christen U, Whitton JL. Molecular mimicry, bystander activation, or viral persistence: Infections and autoimmune disease. Clinical Microbiology Reviews 2006; 19: 80-+. Gayo A, Mozo L, Suarez A, Tunon A, Lahoz C, Gutierrez C. Interferon beta-1b treatment modulates TNFalpha and IFNgamma spontaneous gene expression in MS. Neurology 1999; 52: 1764-1770. Gerlach RF, Tanus-Santos JE. Misuse of serum matrix metalloproteinase (MMP)-9 and tissue inhibitor of metalloproteinase (TIMP)-1 as a biomarker in multiple sclerosis. Mult.Scler. 2006; 12: 120. Gilli F, Marnetto F, Caldano M et al. Biological markers of interferon-beta therapy: comparison among interferon-stimulated genes MxA, TRAIL and XAF-1. Mult.Scler. 2006; 12: 47-57. Goebel MU, Czolbe F, Becker H, Janssen OE, Schedlowski M, Limmroth V. Effects of interferon-beta 1a on the hypothalamic-pituitary-adrenal axis, leukocyte distribution and mood states in multiple sclerosis patients: results of a 1-year follow-up study. Eur.Neurol. 2005; 53: 182-187. Gran B, Zhang GX, Yu S et al. IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J.Immunol. 2002; 169: 7104-7110. Harbo HF, Lie BA, Sawcer S et al. Genes in the HLA class I region may contribute to the HLA class II-associated genetic susceptibility to multiple sclerosis. Tissue Antigens 2004; 63: 237247. p 36 Hartung HP, Bar-Or A, Zoukos Y. What do we know about the mechanism of action of diseasemodifying treatments in MS? Journal of Neurology 2004; 251: 12. He X, Janeway CA, Levine M et al. Dual receptor T cells extend the immune repertoire for foreign antigens. Nature Immunology 2002; 3: 127-134. Hilliard B, Wilmen A, Seidel C, Liu TS, Goke R, Chen Y. Roles of TNF-related apoptosisinducing ligand in experimental autoimmune encephalomyelitis. J.Immunol. 2001; 166: 13141319. Huseby ES, Liggitt D, Brabb T, Schnabel B, Ohlen C, Goverman J. A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. Journal of Experimental Medicine 2001; 194: 669-676. Iwakura Y, Ishigame H. The IL-23/IL-17 axis in inflammation. J.Clin.Invest 2006; 116: 12181222. Jacobs LD, Cookfair DL, Rudick RA et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG). Ann.Neurol. 1996; 39: 285-294. Jensen J, Krakauer M, Sellebjerg F. Cytokines and adhesion molecules in multiple sclerosis patients treated with interferon-beta1b. Cytokine 2005; 29: 24-30. Jensen J, Langkilde AR, Fenst C et al. CD4 T cell activation and disease activity at onset of multiple sclerosis. J.Neuroimmunol. 2004; 149: 202-209. Kallmann BA, Hummel V, Lindenlaub T, Ruprecht K, Toyka KV, Rieckmann P. Cytokineinduced modulation of cellular adhesion to human cerebral endothelial cells is mediated by soluble vascular cell adhesion molecule-1. Brain 2000; 123 ( Pt 4): 687-697. Kantarci OH, Morales Y, Ziemer PA et al. CCR5Delta32 polymorphism effects on CCR5 expression, patterns of immunopathology and disease course in multiple sclerosis. J.Neuroimmunol. 2005; 169: 137-143. p 37 Kantor R, Bakhanashvili M, Achiron A. A mutated CCR5 gene may have favorable prognostic implications in MS. Neurology 2003; 61: 238-240. Khademi M, Wallstrom E, Andersson M, Piehl F, Di Marco R, Olsson T. Reduction of both proand anti-inflammatory cytokines after 6 months of interferon beta-1a treatment of multiple sclerosis. J.Neuroimmunol. 2000; 103: 202-210. Khoury SJ, Guttmann CR, Orav EJ, Kikinis R, Jolesz FA, Weiner HL. Changes in activated T cells in the blood correlate with disease activity in multiple sclerosis. Arch.Neurol. 2000; 57: 1183-1189. Kivisakk P, Cotleur AC, Lee JC, Rudick RA, Ransohoff RM. Interferon-beta 1a does not reduce expression of CCR5 and CXCR3 on circulating T cells. J.Neuroimmunol. 2003; 141: 150-154. Koch-Henriksen N, Bronnumhansen H, Hyllested K. Incidence of Multiple-Sclerosis in Denmark 1948-1982 - A Descriptive Nationwide Study. Neuroepidemiology 1992; 11: 1-10. Kraus J, Ling AK, Hamm S, Voigt K, Oschmann P, Engelhardt B. Interferon-beta stabilizes barrier characteristics of brain endothelial cells in vitro. Ann.Neurol. 2004; 56: 192-205. Kutzelnigg A, Lucchinetti CF, Stadelmann C et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005; 128: 2705-2712. Leppert D, Waubant E, Burk MR, Oksenberg JR, Hauser SL. Interferon beta-1b inhibits gelatinase secretion and in vitro migration of human T cells: a possible mechanism for treatment efficacy in multiple sclerosis. Ann.Neurol. 1996; 40: 846-852. Liu Z, Pelfrey CM, Cotleur A, Lee JC, Rudick RA. Immunomodulatory effects of interferon beta-1a in multiple sclerosis. J.Neuroimmunol. 2001; 112: 153-162. Loetscher P, Uguccioni M, Bordoli L et al. CCR5 is characteristic of Th1 lymphocytes. Nature 1998; 391: 344-345. Lovett-Racke AE, Trotter JL, Lauber J, Perrin PJ, June CH, Racke MK. Decreased dependence of myelin basic protein-reactive T cells on CD28-mediated costimulation in multiple sclerosis p 38 patients - A marker of activated/memory T cells. Journal of Clinical Investigation 1998; 101: 725-730. Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: Results of an international survey. Neurology 1996; 46: 907-911. Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Annals of Neurology 2000; 47: 707-717. Lunemann JD, Waiczies S, Ehrlich S et al. Death ligand TRAIL induces no apoptosis but inhibits activation of human (auto)antigen-specific T cells. J.Immunol. 2002; 168: 4881-4888. Madsen LS, Andersson EC, Jansson L et al. A humanized model for multiple sclerosis using HLA-DR2 and a human T-cell receptor. Nature Genetics 1999; 23: 343-347. Mahad DJ, Lawry J, Howell SJ, Woodroofe MN. Longitudinal study of chemokine receptor expression on peripheral lymphocytes in multiple sclerosis: CXCR3 upregulation is associated with relapse. Mult.Scler. 2003; 9: 189-198. Makhlouf K, Weiner HL, Khoury SJ. Increased percentage of IL-12+ monocytes in the blood correlates with the presence of active MRI lesions in MS. J.Neuroimmunol. 2001; 119: 145-149. Matsui M, Araya SI, Wang HY, Matsushima K, Saida T. Immunomonitoring measures in relapsing-remitting multiple sclerosis. J.Neuroimmunol. 2004; 148: 192-199. Mirowska D, Skierski J, Paz A et al. Changes of percentages in immune cells phenotypes and cytokines production during two-year IFN-beta-1a treatment in multiple sclerosis patients. J.Neurol. 2003; 250: 1229-1236. Misu T, Onodera H, Fujihara K et al. Chemokine receptor expression on T cells in blood and cerebrospinal fluid at relapse and remission of multiple sclerosis: imbalance of Th1/Th2associated chemokine signaling. J.Neuroimmunol. 2001; 114: 207-212. Modin H, Olsson W, Hillert J, Masterman T. Modes of action of HLA-DR susceptibility specificities in multiple sclerosis. American Journal of Human Genetics 2004; 74: 1321-1322. p 39 Muraro PA, Leist T, Bielekova B, McFarland HF. VLA-4/CD49d downregulated on primed T lymphocytes during interferon-beta therapy in multiple sclerosis. J.Neuroimmunol. 2000; 111: 186-194. Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat.Rev.Immunol. 2002; 2: 933-944. Musette P, Benveniste O, Lim A et al. The pattern of production of cytokine mRNAs is markedly altered at the onset of multiple sclerosis. Res.Immunol. 1996; 147: 435-441. Nakajima H, Fukuda K, Doi Y et al. Expression of TH1/TH2-related chemokine receptors on peripheral T cells and correlation with clinical disease activity in patients with multiple sclerosis. Eur.Neurol. 2004; 52: 162-168. Oksenberg JR, Barcellos LF, Cree BAC et al. Mapping multiple sclerosis susceptibility to the HLA-DR locus in African Americans. American Journal of Human Genetics 2004; 74: 160-167. Pantano P, Mainero C, Lenzi D et al. A longitudinal fMRI study on motor activity in patients with multiple sclerosis. Brain 2005; 128: 2146-2153. Polman CH, O'Connor PW, Havrdova E et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N.Engl.J.Med. 2006; 354: 899-910. PRISMS Study Group. Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group. Lancet 1998; 352: 14981504. Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat.Rev.Immunol. 2003; 3: 569-581. Rep MH, Schrijver HM, van Lopik T et al. Interferon (IFN)-beta treatment enhances CD95 and interleukin 10 expression but reduces interferon-gamma producing T cells in MS patients. J.Neuroimmunol. 1999; 96: 92-100. p 40 Rieckmann P, Albrecht M, Kitze B et al. Tumor necrosis factor-alpha messenger RNA expression in patients with relapsing-remitting multiple sclerosis is associated with disease activity. Ann.Neurol. 1995; 37: 82-88. Rieckmann P, Albrecht M, Kitze B et al. Cytokine mRNA levels in mononuclear blood cells from patients with multiple sclerosis. Neurology 1994; 44: 1523-1526. Rudick RA, Ransohoff RM, Lee JC et al. In vivo effects of interferon beta-1a on immunosuppressive cytokines in multiple sclerosis. Neurology 1998; 50: 1294-1300. Sellebjerg F, Madsen HO, Jensen CV, Jensen J, Garred P. CCR5 delta32, matrix metalloproteinase-9 and disease activity in multiple sclerosis. J.Neuroimmunol. 2000; 102: 98106. Sellebjerg F, Sorensen TL. Chemokines and matrix metalloproteinase-9 in leukocyte recruitment to the central nervous system. Brain Res.Bull. 2003; 61: 347-355. Silversides JA, Heggarty SV, McDonnell GV, Hawkins SA, Graham CA. Influence of CCR5 delta32 polymorphism on multiple sclerosis susceptibility and disease course. Mult.Scler. 2004; 10: 149-152. Sindern E, Patzold T, Ossege LM, Gisevius A, Malin JP. Expression of chemokine receptor CXCR3 on cerebrospinal fluid T-cells is related to active MRI lesion appearance in patients with relapsing-remitting multiple sclerosis. J.Neuroimmunol. 2002; 131: 186-190. Song K, Chen Y, Goke R et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is an inhibitor of autoimmune inflammation and cell cycle progression. J.Exp.Med. 2000; 191: 1095-1104. Sorensen TL. Targeting the chemokine receptor CXCR3 and its ligand CXCL10 in the central nervous system: potential therapy for inflammatory demyelinating disease? Curr.Neurovasc.Res. 2004; 1: 183-190. Sorensen TL, Sellebjerg F. Selective suppression of chemokine receptor CXCR3 expression by interferon-beta1a in multiple sclerosis. Mult.Scler. 2002; 8: 104-107. p 41 Sorensen TL, Tani M, Jensen J et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J.Clin.Invest 1999; 103: 807-815. Stuve O, Dooley NP, Uhm JH et al. Interferon beta-1b decreases the migration of T lymphocytes in vitro: effects on matrix metalloproteinase-9. Ann.Neurol. 1996; 40: 853-863. Sun DM, Whitaker JN, Huang ZG et al. Myelin antigen-specific CD8(+) T cells are encephalitogenic and produce severe disease in C57BL/6 mice. Journal of Immunology 2001; 166: 7579-7587. Svejgaard A, Platz P, Ryder LP. HLA and disease 1982--a survey. Immunol.Rev. 1983; 70: 193218. Taylor A, Verhagen J, Blaser K, Akdis M, Akdis CA. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: the role of T regulatory cells. Immunology 2006; 117: 433-442. The IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebocontrolled trial. The IFNB Multiple Sclerosis Study Group. Neurology 1993; 43: 655-661. Trebst C, Ransohoff RM. Investigating chemokines and chemokine receptors in patients with multiple sclerosis: opportunities and challenges. Arch.Neurol. 2001; 58: 1975-1980. Vaknin-Dembinsky A, Balashov K, Weiner HL. IL-23 is increased in dendritic cells in multiple sclerosis and down-regulation of IL-23 by antisense oligos increases dendritic cell IL-10 production. J.Immunol. 2006; 176: 7768-7774. Wandinger KP, Lunemann JD, Wengert O et al. TNF-related apoptosis inducing ligand (TRAIL) as a potential response marker for interferon-beta treatment in multiple sclerosis. Lancet 2003; 361: 2036-2043. Wandinger KP, Sturzebecher CS, Bielekova B et al. Complex immunomodulatory effects of interferon-beta in multiple sclerosis include the upregulation of T helper 1-associated marker genes. Ann.Neurol. 2001; 50: 349-357. p 42 Wiley SR, Schooley K, Smolak PJ et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995; 3: 673-682. Wucherpfennig KW. Mechanisms for the induction of autoimmunity by infectious agents. Journal of Clinical Investigation 2001; 108: 1097-1104. Yong VW, Chabot S, Stuve O, Williams G. Interferon beta in the treatment of multiple sclerosis: mechanisms of action. Neurology 1998; 51: 682-689. Zang YC, Halder JB, Samanta AK, Hong J, Rivera VM, Zhang JZ. Regulation of chemokine receptor CCR5 and production of RANTES and MIP-1alpha by interferon-beta. J.Neuroimmunol. 2001; 112: 174-180. Zhang J, Hutton G, Zang Y. A comparison of the mechanisms of action of interferon beta and glatiramer acetate in the treatment of multiple sclerosis. Clin.Ther. 2002; 24: 1998-2021. Zhang JW, Markovicplese S, Lacet B, Raus J, Weiner HL, Hafler DA. Increased Frequency of Interleukin 2-Responsive T-Cells Specific for Myelin Basic-Protein and Proteolipid Protein in Peripheral-Blood and Cerebrospinal-Fluid of Patients with Multiple-Sclerosis. Journal of Experimental Medicine 1994; 179: 973-984. p 43 9. Summary The PhD dissertation emanated from the Danish MS Research Centre, Rigshosptalet, Copenhagen. Multiple sclerosis (MS) is an inflammatory disease of the CNS. Inflammatory responses by T helper (Th)-lymphocytes are characterised by distinct cytokine expression profiles. In MS, activated Th1-lymphocytes produce proinflammatory cytokines, which induce pathogenic effector cells. Recently, another Th subset relevant to MS has been identified. This is termed Th17 and is partly induced by IL-23. T-cells respond to chemotactic cytokines, termed chemokines, in order to migrate towards sites of inflammation or secondary lymphatic organs. Chemokine receptors are differentially expressed in T cells in blood and cerebrospinal fluid, indicating their role for in T-cell-recruitment to the CNS. Interferon (IFN)-beta is a first-line treatment for MS. The mechanism of action is unclear, but probably includes changes in lymphocyte activation, cytokine secretion, and trafficking. The aim of the studies was to shed more light on T-cell immunology in MS and IFN-beta treatment, as well as identifying putative biomarkers of treatment response and/or disease activity. In one study we identified a Th-cell subset of special interest in MS. This subset expressed CD45R0 and high levels of CD26 as well as a number of activation markers consistent with a phenotype of activated Th1 effector cells. The number of circulating CD45R0+CD26high cells correlated with clinical MS disease severity. IFN-beta treatment had some effects on the expression of apoptosis-related molecules, but no dramatic effects were observed. In a study of chemokines and chemokine receptors we found lower expression of the Th2-related chemokine receptor CCR4 in untreated MS patients compared with healthy controls. IFN-beta therapy decreased expression of the Th1-related CXCR3 as an early effect, while later effects included increased surface expression of CCR4, CCR5, and CCR7. Plasma concentrations of CXCL10 were also increased shortly after an IFN-beta-injection. A study of cytokine mRNA expression revealed increased IL-10 and IL-23 mRNA in MS patients with active disease (not having an acute exacerbation). IFN-beta therapy markedly increased IL-10 mRNA while decreasing IL-23 mRNA expression. These effects were seen as early effects, and tapered quickly after an IFN-beta-injection. No shift towards a Th2 cytokine mRNA expression pattern was seen during IFN-beta therapy. p 44 In conclusion, we have identified a subset of memory CD4+ lymphocytes which may be of special interest in the search for a surrogate marker of disease severity and, possibly, the risk of imminent clinical relapse in MS. Similarly, CXCL10, IL-10 and IL-23 mRNA expression should be evaluated as putative biomarkers of disease activity and treatment response. p 45 10. Dansk resumé PhD afhandlingen udgår fra Dansk Multipel Sclerose Center, Rigshospitalet. Multipel sclerose (MS) er en inflammatorisk sygdom i CNS. Et inflammatorisk respons af Thjælper (Th) lymfocytter er karakteriseret ved en distinkt cytokin ekspression. I MS producerer Th1 lymfocytter pro-inflammatoriske cytokiner, som inducerer patogene effektorceller. For nylig er et andet Th subset af relevans for MS identificeret. Dette kaldes Th17, og induceres bl.a. af IL-23. T celler responderer på kemotaktiske cytokiner, kaldet kemokiner, for at kunne migrere mod områder med inflammation eller sekundære lymfoide organer. Kemokinreceptorer er differentieret udtrykt i T celler i blod og spinalvæske, hvilket antyder deres rolle for T celle rekruttering til CNS. Interferon (IFN)-beta er en førstevalgsbehandling af MS. Virkningsmekanismen er uklar, men inkluderer formentlig ændringer i lymfocytaktivering, –cytokinsekretion og – migration. Formålet med studierne var at udforske T celler immunologien ved MS og under IFN-beta behandling. Desuden at identificere potentielle biomarkører for behandlingsrespons og/eller sygdomsaktivitet. I et studium identificeredes et Th celle subset af særlig relevans for MS. Dette subset udtrykker CD45R0 og CD26 i tillæg til aktiveringsmarkører, foreneligt med en fænotype af aktiverede Th1 effektor celler. Antallet af cirkulerende CD45R0+CD26high celler korrelerede med klinisk MS sygdomssværhedsgrad. IFN-beta behandling påvirkede ekspressionen af nogle apoptose-relaterede molekyler, men der observeredes ingen markante effekter. Under studier af kemokiner og kemokinreceptorer fandtes lavere ekspression af den Th2-koblede kemokinreceptor, CCR4 i ubehandlede MS patienter i forhold til raske. IFNbeta behandling sænkede ekspressionen af den Th1-koblede CXCR3 som en tidlig effekt, mens protraherede effekter omfattede øget overfladeekspression af CCR4, CCR5 og CCR7. Plasmakoncentrationer af CXCL10 øgedes også kort efter en IFN-beta injektion. Cytokin mRNA kvantificering viste øget IL-10 og IL-23 mRNA i MS patienter med aktiv sygdom (uden attak). IFN-beta behandling øgede IL-10 mRNA betydeligt mens IL-23 mRNA sænkedes. Disse effekter sås som tidlige effekter, og klingede hurtigt af efter injektionen. Der sås ingen skift hen imod en Th2 cytokin mRNA ekspressionsprofil under IFN-beta behandling. p 46 Sammenfattende har vi identificeret et subset af memory Th lymfocytter som kan være af særlig interesse som surrogatmarkør for sygdomssværhedsgrad og muligvis øget sygdomsaktivitet (attakker). Ligeledes bør CXCL10, IL-10 og IL-23 mRNA ekspression evalueres som potentielle biomarkører for sygdomsaktivitet og behandlingsrespons..