Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Drosophila melanogaster wikipedia , lookup
DNA vaccination wikipedia , lookup
Adaptive immune system wikipedia , lookup
Molecular mimicry wikipedia , lookup
Polyclonal B cell response wikipedia , lookup
Cancer immunotherapy wikipedia , lookup
Innate immune system wikipedia , lookup
Adoptive cell transfer wikipedia , lookup
Psychoneuroimmunology wikipedia , lookup
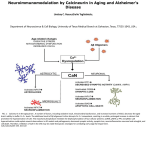
NEWS AND VIEWS Calcium-independent calcineurin regulation Richard V Parry & Carl H June CaM CHP + – CnB nB – FK506 CnA – Csp1 A238L NFAT CnA — P P CABIN MEF 2 No response NFAT Im mmune response gene es CC C.C. – Richard V. Parry and Carl H. June are at the Abramson Family Cancer Research Institute and Department of Pathology and Laboratory Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA. e-mail: [email protected] 2+ Ca – Although intracellular signal transduction is often portrayed as a protein kinase ‘domino effect’, the counterbalancing function of phosphatases, and thus the control of phosphatase activity, is equally relevant to proper regulation of cellular function. Indeed, a notable example of the importance of phosphatases is found in the plague–causing agent Yersinia pestis, which depends on a phosphatase to exert its deadly effect1. In contrast to the many protein kinases that have been discovered and studied in detail, relatively few phosphatases have received similar attention. One prominent exception is found in calcineurin (PP2B), a calcium-responsive enzyme that dephosphorylates nuclear factor of activated T cells (NFAT), thereby promoting its nuclear translocation, and thus occupies a unique niche linking calcium signaling to transcriptional regulation2. In this issue of Nature Immunology, Ryeom and colleagues describe a new level of calcineurin regulation mediated by a group of proteins called calcipressins3. Using calcipressin 1 (Csp1)-deficient mice, they demonstrate Csp1 as an in vivo regulator of calcineurin, and explore its involvement in the acquired immune response. Furthermore, these results have provided more insights about the function of calcineurin itself. Protein phosphatases are subdivided into three classes: serine/threonine specific, tyrosine specific and dual specificity. Based on biochemical parameters, substrate specificity and sensitivity to various inhibitors, serine/threonine protein phosphatases are divided into two main classes. Type I protein phosphatases can be inhibited by two heat-stable proteins, called inhibitor 1 and inhibitor 2. Type II protein phosphatases are insensitive to heat-stable inhibitors and are + © 2003 Nature Publishing Group http://www.nature.com/natureimmunology Calcineurin is negatively regulated by calcipressin 1. Analysis of calcipressin-deficient mice shows that survival of T helper type 1 cells is dependent on calcipressin, demonstrating another function for the regulation of calcineurin activity in T cells. Figure 1 Multiple levels of regulation occur in the calcium-calcineurin-NFAT pathway. Calcineurin (Cn) is activated by the binding of calcineurin A (CnA) to calcineurin B (CnB) and calmodulin (CaM), displacing an auto-inhibitory domain. Dephosphorylation of NFAT promotes its nuclear import. FK506- and FKBPimmunophilin complexes bind CnB, calcipressin (Csp1) binds CnA, and CHP competes with CaM for CnA binding. The viral inhibitor A238L blocks Cn-NFAT interaction, and cabin1 interacts with activated calcineurin in the nucleus, preventing MEF2 activation, in this example. further subdivided into spontaneously active (PP2A), calcium-dependent (PP2B; calcineurin) and magnesium-dependent (PP2C) classes of phosphatases. Calcineurin was first identified as a principal calmodulin-binding protein from brain, and is abundantly expressed in areas of the brain that are vulnerable to stroke, epilepsy and neurodegenerative diseases. Calcineurin is important in inflammation and, in the clinic, immunosuppression can be achieved by administration of cyclosporin A and FK506, which target calcineurin phosphatase activity, blocking T cell activation. Calcineurin is a heterodimer consisting of a 60-kDa catalytic A subunit and a 19-kDa regulatory B subunit. The catalytic subunit shares 40% sequence homology with PP2A but contains a C-terminal region that functions as a calmodulin-binding domain. The catalytic activity of calcineurin is positively regulated by the binding of cal- NATURE IMMUNOLOGY VOLUME 4 NUMBER 9 SEPTEMBER 2003 cium both directly to calcineurin B and as a calcium-calmodulin complex, which displaces an autoinhibitory domain, allowing access of substrates to the catalytic domain. Recent work, particularly in cardiac myocytes, has indicated that calcipressins may negatively regulate calcineurin activity by directly binding CnA4. Calcipressins are a family of proteins derived from three genes. Calcipressin 1 is also known as modulatory calcineurin-interacting protein 1 (MCIP1), Adapt78 and Down syndrome critical region 1 (DSCR1). Calcipressin 2 is variously known as MCIP2, ZAKI-4 and DSCR1-like 1. Calcipressin 3 is also called MCIP3 and DSCR1-like 2. The Human Nomenclature Committee has assigned DSCR designations to these genes; however, for clarity, the Csp1, Csp2 and Csp3 nomenclature will be used here. In a series of in vitro experiments, Ryeom et al. demonstrate that calcipressins bind activated calcineurin 821 © 2003 Nature Publishing Group http://www.nature.com/natureimmunology NEWS AND VIEWS through their C termini and exert a potent inhibitory effect on calcineurin phosphatase activity and calcium-driven nuclear import of NFATc, similar to that seen with immunophilin-cyclosporin A or FK506 complexes. Despite the profound influence of calcineurin on lymphocyte function, the expression and function of calcipressins in T lymphocytes has received scant attention so far. Here, Ryeom et al. confirm the expression of Csp1 in wild-type mouse lymph node CD4 T cells, and the generation of Csp1-deficient mice, achieved by targeting DSCR1 exon 6. As all four reported splice variants of Csp1 encode exon 6, no Csp1 protein is expressed. Despite the absence of Csp1, deficient mice show no apparent abnormalities in lymphocyte development or homeostasis. Catalytic activity of endogenous calcineurin in response to ionomycin, however, was found to be greater in the absence of Csp1, consistent with the proposed inhibitory function of Csp1 (ref. 5). Unexpectedly, the proliferation of CD4 T cells in response to stimulation with antibodies to CD3 and CD28 was impaired in Csp1-deficient cells compared with that of wild-type T cells. Csp1-deficient cells also showed a notable deficit in production of interferon-γ, but not interleukin 4, indicating that T helper type 1 (TH1) cells were specifically affected. Furthermore, analysis of cells after primary stimulation showed low expression of T-bet but normal expression of GATA-3 (TH1- and TH2-specific transcription factors, respectively). These in vitro data were recapitulated in vivo by immunization of mice with 2,4,6-trinitrophenol–keyhole limpet hemocyanin (TNPKLH), an antigen that can induce a mixed TH1-TH2 response. Csp1-deficient mice showed TNP-specific immunoglobulin M (IgM) and IgG1 concentrations comparable to those of wild-type mice, but decreased concentrations of TNP-specific IgG2a. This is taken as further evidence of defective TH1 cytokine production, as the TH1 cytokine interferon-γ promotes switching to IgG2a isotype, whereas the TH2 cytokine interleukin 4 promotes switching to IgG1 and IgE. The defect in the TH1 immune response noted in Csp1-deficient lymphocytes could result from a defect in TH1 differentiation, proliferation or survival. Investigating the mechanism underlying the impaired TH1 response, the authors demonstrated increased apoptosis of Csp1-deficient TH1 cells but not Csp1-deficient TH2 cells. In mice, homeostasis of TH1 cells is achieved by apoptosis as a result of calcineurindependent Fas ligand expression during 822 secondary stimulation6. Therefore, the authors investigated whether aberrant expression of Fas ligand mediated increased cell death after primary stimulation of Csp1-deficient TH1 cells. The authors were able to restore TH1 function in Csp1-deficient cells by carefully titrating cyclosporin A to mimic Csp1-mediated calcineurin inhibition and by blocking cell death in Csp1-deficient TH1 using Fas ligand–neutralizing antibodies, which increased proliferation and restored interferon-γ production in Csp1-deficient cells. Thus, there is compelling evidence that aberrant expression of Fas ligand precipitates the TH1 defect in Csp1-deficient lymphocytes. Finally, the authors propose a hierarchical model for Csp1 alteration of calcineurin dependent transcription. The activation thresholds of several genes were measured in terms of T cell receptor (TCR) signal strength; these thresholds were found to be lower in Csp1-deficient cells. As expression of Fas ligand required the greatest TCR signal strength of the genes examined, the consequence of losing Csp1-mediated calcineurin inhibition maybe to allow aberrant Fas ligand expression after primary stimulation of TH1 cells. Thus the data indicate that Csp1 has a key regulatory function in the calcium-calcineurin-NFAT signaling pathway, altering the transcriptional profile induced by calcium signaling. The present work provides insight into the involvement of calcipressins in T lymphocytes, and thus many questions remain unanswered. As previously described, the calcipressin family is derived from three genes, and the expression of these genes and their splice variants has not been fully described in T lymphocytes. Although Ryeom et al. demonstrate that Csp1, Csp2 and Csp3 are all capable of blocking calcineurin activity, these molecules are presumably not functionally redundant, and the effect of Csp2 and Csp3 deficiency remains to be described. The authors show that the C termini of calcipressins are sufficient for calcineurin inhibition; however, different calcipressins may have distinct functions beyond calcineurin regulation, and thus their deficiency may demonstrate unforeseen phenotypes. The mechanisms controlling tissue-specific expression probably differ for individual calcipressins; indeed, work in other cell types has indicated that the expression of Csp1 and Csp2 is independently controlled by calcineurin and thyroid hormone, respectively7. As Csp1 expression is increased by calcineurin activity, this may form the basis of a feedback inhibition loop, which could serve to protect T cells from high or sustained intracellular calcium when antigen excess is encountered, as has been suggested for neurons5. Precedence has been set for such a system in T lymphocytes by the molecule A20, which is induced by NF-κB, but promotes NF-κB nuclear export. The upstream signaling mechanisms that regulate calcipressin activity remain a chief unexplored question. Csp1 encodes a highly conserved central serine-proline repeat sequence that is subject to phosphorylation by glycogen synthase kinase (GSK) 3 and mitogen-activated protein (MAP) kinase, and is itself a substrate for calcineurin. Phosphorylation status will likely modulate calcipressin activity, and as both GSK3 and MAP kinase are regulated at least in part by costimulatory signals, the influence of costimulation on calcipressin activity may be substantial in T lymphocytes. Positive costimulatory signals delivered by CD28 and ICOS mediate increased intracellular calcium8. In contrast, negative costimulatory molecules such as CTLA-4, PD-1 and the recently described BTLA9 exert an inhibitory effect over interleukin 2 transcription, and it is possible that regulation of calcipressin activity may help them achieve this. Although work so far has focused on the inhibitory function of calcipressins, it has been suggested that Csp1 may suppress or enhance the activity of calcineurin, depending on the context of the cellular stimulus10. The function of calcipressin may therefore be more complex than its merely acting as a calcineurin inhibitor. Furthermore, there are several classes of calcineurin inhibitors in addition to calcipressins (Fig. 1). The calcium-dependent activation of calcineurin is prevented by calcineurin B homologous protein, which competes with calcineurin B for binding to calcineurin A11. Cabin 1 (also call Cain) is a mainly nuclear, noncompetitive inhibitor that interacts with activated calcineurin12. Of particular interest to immunologists is the A238L protein encoded by the African swine fever virus. This protein shares sequence similarity with the calcineurin interaction domain of NFAT family members, and thus can mediate immunosuppression and permit the virus to evade the host immune response13. A further appreciation of the signals that regulate these calcineurin inhibitors may yield clues as to their functions, and their potential for manipulation in lymphocytes. The mechanisms by which diverse cell types generate appropriate and specific responses to changes in intracellular calcium VOLUME 4 NUMBER 9 SEPTEMBER 2003 NATURE IMMUNOLOGY © 2003 Nature Publishing Group http://www.nature.com/natureimmunology NEWS AND VIEWS concentration has been an enduring biological question. Answers probably lie in tissuespecific regulatory molecules, and in this respect the emergence of calcipressins as regulators of calcineurin function is a promising development. This demonstration of the regulatory function of calcipressins in T lymphocytes advances our knowledge of the immune system, adding a subtlety to calcineurin function. The phenotype of Csp1 deficiency in T cells shows the potential involvement that calcineurin regulatory proteins have in the therapeutic manipulation of the immune system. 1. Guan, K.L. & Dixon, J.E. Science 249, 553–556 (1990). 2. Clipstone, N.A. & Crabtree, G.R. Nature 357, 695–697 (1992). 3. Ryeom, S., Greenwald, R.J., Sharpe, A.H. & McKeon, F. Nat. Immunol. 4, 874–881 (2003). 4. Rothermel, B.A., Vega, R.B. & Williams, R.S. Trends Cardiovasc. Med. 13, 15–21 (2003). 5. Ermak, G., Harris, C.D. & Davies, K.J. FASEB J. 16, 814–824 (2002). 6. Zhang, X. et al. Unequal death in T helper cell (Th)1 and Th2 effectors: Th1, but not Th2, effectors undergo rapid Fas/FasL-mediated apoptosis. J. Exp. Med. 185, 1837–1849 (1997). 7. Yang, J. et al. Circ. Res. 87, E61–E68 (2000). 8. Parry, R.V., Rumbley, C.A., Vandenberghe, L.H., June, C.H. & Riley, J.L. J. Immunol. 171, 166–174 (2003). 9. Watanabe, N. et al. Nat. Immunol. 4, 670–679 (2003). 10. Vega, R.B. et al. Proc. Natl. Acad. Sci. USA 100, 669–674 (2003). 11. Lin, X., Sikkink, R.A., Rusnak, F. & Barber, D.L. J. Biol. Chem. 274, 36125–36131 (1999). 12. Sun, L. et al. Immunity 8, 703–711 (1998). 13. Miskin, J.E., Abrams, C.C., Goatley, L.C. & Dixon, L.K. Science 281, 562–565 (1998). SIGIRR puts the brakes on Toll-like receptors Luke AJ O’Neill Members of the Toll-like receptor–interleukin 1 receptor superfamily signal inflammatory responses. However, a member of this family is now shown to modulate these responses by acting as a negative regulator. The initial phase of host defense against invading microbes involves a family of proteins called Toll-like receptors (TLRs). These proteins are expressed on various cell types, most notably dendritic cells, where they act as primary sensors of microbial products and activate signaling pathways that lead to the induction of immune and inflammatory genes. TLRs belong to a broader family of proteins, which include receptors for the pro-inflammatory cytokines interleukin 1 (IL-1) and IL-18 (ref. 1). Among the bestcharacterized TLRs are TLR4, TLR5 and TLR9, which sense lipopolysaccharide (LPS), flagellin and CpG motifs, respectively. Although these receptors have important functions in host defense, their unrestrained stimulation may be detrimental to the host. Thus, negative regulators of IL-1 receptor (IL-1R), IL-18R and TLRs may be required to modulate their responses. In this issue of Nature Immunology, Wald et al.2 describe an intriguing inhibitor of this receptor superfamily. All members of the TLR–IL-1R superfamily signal inflammation in a very similar way. This is because they all contain a conserved protein sequence in their cytosolic regions, called the Toll–IL-1R (TIR) domain, which activates common signaling pathways, most notably those leading to the activation of the tran- Luke A.J. O’Neill is in the Cytokine Research Group, Department of Biochemistry, Trinity College, Dublin, Ireland. e-mail: [email protected] scription factor NF-κB and stress-activated protein kinases. However, Wald et al. show that an orphan receptor, which has the rather cumbersome but accurate name, single immunoglobulin IL-1R–related protein (SIGIRR)3, is an inhibitory member of this receptor superfamily. SIGIRR seems to temper cellular activation by IL-1, LPS and probably other activators of receptors in the TLR–IL-1R superfamily, such that the biological outcome will be the result of a balance between activation by a receptor and dampening by SIGIRR. SIGIRR therefore acts as a ‘brake’ on the TLR system, which may be essential for regulating the detrimental effects of innate immunity, as occurs in sepsis and chronic inflammation. The TLR–IL-1R superfamily can be divided into three subgroups1. The first contains extracellular immunoglobulin (Ig) domains and includes IL-1RI. The second is the TLRs, which lack Ig domains, but have extracellular leucine-rich repeats; recent years have seen tremendous progress in determining their function. The third subgroup consists of upstream adapter molecules, including MyD88, MyD88 adapter-like (Mal, also known as TIRAP) and TIR domain–containing adaptor–inducing interferon-β (IFN-β; TRIF, also known as TICAM-1). These adapters are recruited to receptor TIR domains and initiate signalling processes through IL-1R–associated kinases (of which there are four) and the adaptor molecule TRAF-6, which leads to activation of four protein kinase cascades, culminating in the activation of NF-κB and kinases p38, JNK and p42/p44 MAP kinase1. These molecules in turn promote the production of many NATURE IMMUNOLOGY VOLUME 4 NUMBER 9 SEPTEMBER 2003 proinflammatory proteins and enhance immune reactivity. Recent evidence indicates differences in adapter usage, such that although almost all the receptors recruit MyD88, only some receptors use Mal and TRIF, leading to specificity in outcome4. The best example is TRIF usage by TLR3 and TLR4, which leads to the activation of IFN-regulatory factor 3 and the induction of IFN-β5,6. To some extent, because there has been much more progress made in the understanding of the TLR and adapter subgroups, the Ig subgroup has been neglected. Most members in this subgroup remain as orphan receptors of unknown function, the exceptions being IL-1R and IL-18R and their respective accessory proteins, and IL-1Rrp2, which may be a receptor for IL-1F9, a ‘paralog’ of IL-1 (ref. 7). Five other IL-1 paralogs occur in humans and it seems likely that they will be ligands or antagonists for the orphan receptors8. Defining a function for SIGIRR therefore assigns a function to one of the orphans; it is of considerable interest that this function is inhibitory for other members of the protein superfamily. Wald et al. show that SIGIRR is expressed in various mouse tissues, including epithelial cells in the kidney, and is highly expressed in the colon but less so in spleen cells. Bone marrow–derived macrophages did not express SIGIRR. Because LPS down-regulates SIGIRR expression in epithelial cells, this indicates that SIGIRR might be inhibitory, consistent with the fact that SIGIRR has a TIR domain that lacks two amino acids essential for signaling by IL-1RI. To test this possibility, the authors over-expressed SIGIRR in Jurkat 823