Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
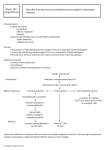
Noah W. Palm Ruslan Medzhitov Pattern recognition receptors and control of adaptive immunity Authors’ address Noah W. Palm1, Ruslan Medzhitov1 1 Howard Hughes Medical Institute and Department of Immunobiology, Yale University, School of Medicine, New Haven, CT, USA. Summary: The mammalian immune system effectively fights infection through the cooperation of two connected systems, innate and adaptive immunity. Germ-line encoded pattern recognition receptors (PRRs) of the innate immune system sense the presence of infection and activate innate immunity. Some PRRs also induce signals that lead to the activation of adaptive immunity. Adaptive immunity is controlled by PRRinduced signals at multiple checkpoints dictating the initiation of a response, the type of response, the magnitude and duration of the response, and the production of long-term memory. PRRs thus instruct the adaptive immune system on when and how to best respond to a particular infection. In this review, we discuss the roles of various PRRs in control of adaptive immunity. Correspondence to: Ruslan Medzhitov Howard Hughes Medical Institute and Department of Immunobiology Yale University, School of Medicine 300 Cedar Street New Haven, CT 06510, USA Tel.: +1 203 785 7541 Fax: +1 203 785 4461 e-mail: [email protected] Keywords: Toll-like receptors ⁄ pattern recognition receptors, T cells, inflammation, cell activation Introduction Immunological Reviews 2009 Vol. 227: 221–233 Printed in Singapore. All rights reserved 2009 The Authors Journal compilation 2009 Blackwell Munksgaard Immunological Reviews 0105-2896 All living organisms, from bacteria through humans, have evolved strategies to counter parasitic infections (1). In higher organisms, the varied and numerous strategies involved in defense from parasitic microbes are collectively referred to as the immune system. The mammalian immune system consists of two interrelated arms—the evolutionarily ancient and immediate innate immune system, and the highly specific, but temporally delayed, adaptive immune system. The combination of innate and adaptive immunity enables the mammalian immune system to recognize and eliminate invading pathogens with maximal efficacy and minimal damage to self, as well as to provide protection from re-infection with the same pathogen. The innate and adaptive immune systems use two fundamentally different strategies to recognize microbial invaders—specifically, the innate immune system detects infection using a limited number of germ-line encoded receptors that recognize molecular structures unique to classes of infectious microbes, while the adaptive immune system uses randomly generated, clonally expressed, highly specific receptors of seemingly limitless specificity (2, 3). It is the combination of these two strategies of recognition that makes the mammalian immune system highly efficacious. 2009 The Authors • Journal compilation 2009 Blackwell Munksgaard • Immunological Reviews 227/2009 221 Palm & Medzhitov Æ Innate instruction of adaptive immunity The mammalian immune system evolved to maximize host defense while minimizing damage to the host tissues. The innate immune system employs a variety of highly efficient immune effectors to combat infection; however, the efficacy of these effectors is limited by the collateral damage, or immunopathology, they can cause. Thus, the efficacy of the innate immune system is limited by the level of immunopathology that can be tolerated. The adaptive immune system increases the potential efficacy of immunity partially by minimizing collateral damage by focusing immune defense in an antigen-specific manner using highly specific antigen receptors. However, by using randomly generated receptors that cannot reliably distinguish self from non-self, adaptive immunity creates another set of opposing forces that must be balanced: the ability of the adaptive immune system to respond to pathogenic non-self must be maximized, while the possibility of responses to self and ⁄ or innocuous non-self must be minimized. The innate and adaptive immune systems are ideal partners because the differing strategies that they use to recognize infection possess complementary strengths and weaknesses. Because the innate immune system uses germ-line encoded receptors to recognize invariant features typical of classes of microbes, it is highly efficient at distinguishing self from non-self (4). However, the non-clonal mechanism of activation of innate immune effectors has the potential to cause significant collateral damage to self-tissues resulting in immunopathology. Adaptive immunity, because it uses random, diverse, clonally expressed receptors produced via somatic recombination, is highly effective in specifically targeting immune responses towards the infection while sparing the surrounding uninfected tissue. However, cells of the adaptive immune system cannot by themselves reliably determine the origin of the antigen they are specific for. By working together, innate and adaptive immunity can both effectively target immune effectors and reliably distinguish self from non-self. A conceptual framework for the current understanding of the functioning of the innate immune system and its control of adaptive immunity was proposed by the late Charles Janeway Jr nearly 20 years ago (4). Janeway’s hypothesis was essentially as follows: the adaptive immune system, because of the use of randomly generated receptors for antigen recognition, cannot reliably distinguish between self and non-self. Therefore, adaptive immune cells must be instructed as to the origin of an antigen by a system that can determine, with high fidelity, whether an antigen is derived from self, infectious (i.e. microbial) non-self, or innocuous (i.e. non-infectious and non-microbial) non-self. Janeway suggested that the 222 evolutionarily ancient, and at that point severely understudied, innate immune system might be able to provide such instruction. Furthermore, he proposed a concrete mechanism by which the innate immune system could sense the presence of infection and relay its conclusions to the adaptive immune system. Janeway posited that the innate immune system would sense the presence of infection via recognition of conserved microbial pathogen-associated molecular patterns (PAMPs), by germ-line encoded receptors he dubbed pattern recognition receptors (PRRs). These PAMPs would possess certain qualities to be effective. First, they would have to be unique to microbes and absent from eukaryotic cells so that they would accurately signal infection. Second, they would be common to a broad class of microbes so that a limited number of germ-line encoded receptors could detect all infections. Third, they would be essential for the life of the microbe so that their detection could not be easily eliminated via mutation. Maybe most importantly, Janeway also predicted that recognition of infection by PRRs on cells of the innate immune system would lead to the induction of signals involved in activation of the adaptive immune system and thus would result in initiation of adaptive immunity. This conceptual framework has now been proven to be correct, although new advances naturally require further development of the theory. Adaptive immunity: advantages and dangers Antigen receptors of the adaptive immune system offer a number of unique advantages because of their ability to recognize nearly any antigen and their clonal expression (3). First, adaptive immune responses possess exquisite specificity and, thus, can specifically target the immune response; therefore, adaptive immunity can maximize the efficacy of the immune response while minimizing unnecessary collateral damage. Second, because of clonal expression and selection, adaptive immunity endows the immune system with a mechanism by which to remember previous infections and, thus, provide protection from future infection with the same pathogen. Because randomly generated antigen receptors cannot determine the origin of the antigen, they can potentially activate the immune response against self-antigens, leading to autoimmunity, or innocuous non-self antigens, leading to allergy. Negative selection of autoreactive lymphocytes in the thymus provides an important, albeit incomplete, solution to the self ⁄ non-self discrimination problem. However, negative selection is incomplete in the sense that it does not eliminate all self-reactive specificities and it cannot account for the 2009 The Authors • Journal compilation 2009 Blackwell Munksgaard • Immunological Reviews 227/2009 Palm & Medzhitov Æ Innate instruction of adaptive immunity discrimination between different sources of non-self antigens (for example, microbial versus food antigens). Thus, despite central tolerance, opportunities for inappropriate adaptive immune responses abound, and other mechanisms to prevent inappropriate responses also exist (5). Peripheral tolerance consists of a variety of mechanisms that prevent inappropriate responses by autoreactive lymphocytes that escape negative selection and lymphocytes specific for innocuous non-self antigens. One aspect of peripheral tolerance is the inactivation of T cells that see their antigen in the absence of signals indicating the presence of infection; these cells are thus rendered permanently unresponsive or anergic (6). Another aspect of peripheral tolerance is enforced by regulatory T cells, which suppress, via various mechanisms, inappropriate adaptive immune responses to self or innocuous non-self (7). Checkpoints in control of adaptive immunity The adaptive immune response is a multistep process where different types of decisions are made before the onset of every subsequent stage. These checkpoints control different aspects of the adaptive immune responses and integrate information provided by various signals induced by infection through PRRs (Fig. 1). The major checkpoints determine the origin of the antigen, the type of infection (pathogen class), the extent and duration of infection, and finally, the requirement for immediate defense or future defense. The first checkpoint is controlled by the signals that indicate the origin of the antigen. This is presumably a binary, yes or no, decision that determines whether to initiate the response. Dendritic cells (DCs), as well as other cell types of the innate immune system, upon activation by PRRs, produce a variety of signals that couple the identity of the antigen with its microbial origin (8). The coupling mechanisms often rely on physical association between an antigen and a PAMP. For example, attachment of the C3dg fragment to an antigen ‘flags’ the antigen as foreign, or microbial, and directs efficient antibody responses against the antigen (9). Similarly, co-occurrence of antigens and Toll-like receptor (TLR) ligands in the same phagosome (which normally only happens if the two are physically associated, for example as parts of the same bacterial cell), signals the microbial origin of the antigen and promotes selection of microbial antigens for major histocompatibility complex class II presentation by DCs (10–12). In general, however, the exact mechanisms involved in coupling antigens with PRR ligands are not entirely understood. Signals produced by PRRs that indicate the origin of the antigen to T cells are also not well defined. Although the costimulatory signals, CD80 and CD86, are commonly thought to be critical, by themselves they are clearly not sufficient to induce T-cell responses (13, 14). The second checkpoint controls the type of adaptive immune response induced. To be effective in combating infection, adaptive responses are tailored to the class of infection; for example, T-helper 1 (Th1) responses are induced PRR-Induced Signals Fig. 1. PRR-mediated control of checkpoints of adaptive immunity. Pattern recognition receptors (PRRs) detect the presence and features of an infection and PRR-induced signals control adaptive immunity accordingly at various checkpoints to insure a productive adaptive response. PRR-mediated determination of whether an antigen is self, innocuous non-self, or non-innocuous non-self leads to control of activation of an adaptive response. PRR-mediated determination of the class of infection leads to control of the type of adaptive response induced. PRR-mediated determination of the level of infection and persistence of an infection leads to control of the magnitude and duration of the adaptive response. Finally, PRR-mediated determination that immediate defense is needed to combat an ongoing infection leads to effector cell generation, while determination that an infection has been cleared and resources can be shuttled towards future defense leads to memory cell generation. A failure to properly regulate immunity at each checkpoint can lead to various immune pathologies including autoimmunity, allergy, failure to protect from infection, and immunopathology. 2009 The Authors • Journal compilation 2009 Blackwell Munksgaard • Immunological Reviews 227/2009 223 Palm & Medzhitov Æ Innate instruction of adaptive immunity in response to intracellular bacteria, Th2 responses are induced in response to helminths, and Th17 responses are induced in response to extracellular bacteria and fungi. PRRs sense the presence of different classes of infection and control the induction of appropriate effector responses through the production of specialized instructive signals. Thus, bacterial infections can induce interleukin-12 (IL-12) production, viral infections trigger type I interferon (IFN) production, and helminthes induce thymic stromal lymphopoietin, IL-4, and IL-13 production. These signals can in turn direct T-cell differentiation into the appropriate effector classes (15). Two related checkpoints in the adaptive immune response control the magnitude and duration of the response, which should correlate with the extent and persistence of infection, respectively. Unlike the origin of the antigen or the class of pathogen, the magnitude and duration of a response are graded functions. They can be ‘read-out’ by PRRs that can sense the extent and the persistence of infection because infection is the source of PRR ligands. Aside from a few model systems, little is known about how the magnitude of the adaptive immune response is controlled. Since the magnitude of an immune response is dependent upon the extent of infection, presumably it is controlled by a signal that is proportional to the level of infection. The duration of response may be controlled by similar signals that must be produced only in the presence of infection. Indeed, in the case of Listeria infection, the initial infectious dose and antigen dose are critical for determining the magnitude of the CD8+ T-cell response, whereas the persistence of infection determines the duration of the response (16). Thus, both antigens and PAMPs may be sensed to co-ordinately control the magnitude and duration of the adaptive immune response. In addition, magnitude and duration are also controlled by negative regulators of the response, such as the negative costimulatory molecule cytotoxic T-lymphocyte antigen-4. In addition to the checkpoints that control the primary immune response, there is a checkpoint that controls the generation and activation of a memory response. This checkpoint is also regulated by PRRs. Recent data on TLR-induced adaptive responses have demonstrated that, under certain conditions, a primary CD4+ T-cell response can be induced that does not lead to T-cell memory (13). These data suggested the existence of a PRR-induced signal that is necessary for memory generation, but dispensable (at least under certain experimental conditions) for induction of a primary response. The identity of this signal is currently unknown. In another study, TLR-mediated activation of memory B cells 224 was shown to elicit a memory antibody response, indicating that the recall response is also regulated by PRRs (17). Memory generation is also controlled by the perceived need for immediate defense (effector cells) during ongoing infection versus future defense (memory cells) after the infection has been cleared; this also is controlled by PRRs. PRRs and control of adaptive immunity All PRRs can detect the presence and type of microbial infection and activate the appropriate innate immune response (18). Some PRRs can also control adaptive immunity accordingly, particularly at the various checkpoints outlined above. This instruction of adaptive immunity occurs largely through triggering the maturation of DCs from highly phagocytic, weakly immunogenic, tissue-resident cells into weakly phagocytic, highly immunogenic, lymph node-homing cells that are competent to induce tailored T-cell responses to non-self antigens acquired in the periphery (8, 19). Not all PRRs are equal in terms of their ability to trigger the adaptive immune response. Indeed, while some PRRs (e.g. TLRs) are sufficient to induce both T- and B-cell responses, other PRRs (e.g. the mannose receptor and scavenger receptors) are not competent to induce adaptive immunity by themselves (8). Presumably, the ability and sufficiency of a given PRR to control adaptive immunity at each of the checkpoints discussed above should correlate with the ability of that particular PRR to accurately detect the presence, extent, and duration of infection, and the microbial origin of the antigens, as well as to effectively relay this information to the adaptive immune system. In the next sections we discuss what is currently known about control of adaptive immunity by different classes of PRRs (Table 1). For this purpose, two classes of PRRs, transmembrane and cytosolic, are discussed separately. Transmembrane PRRs Transmembrane PRRs recognize PAMPs in the extracellular space and ⁄ or in phagosomes or endosomes. Here, we discuss members of two families of transmembrane PRRs—the TLRs and the C-type lectin Dectin-1. TLRs TLRs are the canonical PRRs that fulfill all of the predicted qualities of a PRR that links innate and adaptive immunity—that is, TLRs sense infection through the recognition of PAMPs and induce appropriately tailored innate and adaptive 2009 The Authors • Journal compilation 2009 Blackwell Munksgaard • Immunological Reviews 227/2009 Palm & Medzhitov Æ Innate instruction of adaptive immunity Table 1. Transmembrane and cytosolic PRRs and their connection to adaptive immunity PRR Ligands recognized Microbes recognized Adaptive response induced References Gram-positive and Gram-negative bacteria, DNA and RNA viruses, fungi, protozoa Fungi Sufficient to induce Th1, antibody (particularly IgG2), and CD8+ T-cell responses (Reviewed in 8, 20) Sufficient to induce Th17 and antibody responses (31, 32) Cytosolic bacterial cell wall components, peptidoglycans Intracellular bacteria (48, 50) Pathogenic bacteria ISD sensor Potassium efflux, LPS plus ATP, pore-forming toxins, bacterial secretion systems Cytosolic DNA RIG-I ⁄ MDA5 Cytosolic RNA RNA viruses Sufficient to induce Th2 and antibody responses; potentiates Th1, Th2, Th17, and antibody responses initiated by TLR; may favor Th17 responses Required for robust T cell-dependent hypersensitivity; may potentiate Th2 driven antibody responses Sufficient to induce CD4+ T-cell and antibody responses (in hematopoietic cells); sufficient to induce CD8+ T-cell responses (in non-hematopoietic cells) Sufficient to induce CD8+ T-cell responses; insufficient to induce CD4+ T-cell and antibody responses Transmembrane PRRs TLRs Bacterial cell wall components, viral nucleic acids in endosomes, etc. Dectin-1 Cytosolic PRRs Nod1, Nod2 NALP3 Fungal cell wall components, b-glucan DNA viruses, retroviruses (53–58) (64) (69) PRRs, pattern recognition receptors; TLR, Toll-like receptor; Nod, nucleotide-binding oligomerization domain; NALP3, Nacht domain-, LRR-, and PYDcontaining protein 3; ISD, interferon stimulatory DNA; RIG-I, retinoic acid-inducible gene I; MDA5, melanoma differentiation-associated gene 5; LPS, lipopolysaccharide; ATP, adenosine triphosphate; Th, T-helper; IgG2, immunoglobulin G2. immune responses (8, 20). TLRs have been shown to be sufficient to control adaptive immunity at all checkpoints leading to a tailored adaptive response, characterized by Th1 induction, immunoglobulin G2c (IgG2c) production, CD8+ T-cell induction, and protection from re-infection (13, 21, 22). TLRs have also been shown to be critical for the induction of adaptive immunity in response to various immunizations and infections (20, 23). The critical roles of TLRs in activation of adaptive immune responses have been well documented and extensively reviewed elsewhere (8, 20). TLRs have been shown to control adaptive immune responses at multiple levels, including control of antigen uptake (24) and antigen selection for presentation in DCs (12), control of DC maturation and cytokine production (8, 13), control of naive T-cell susceptibility to suppression by regulatory T cells (Tregs) (25), and control of survival of activated T cells (26). TLRs can also control B-cell responses to T-dependent and T-independent antigens (21, 27, 28), as well as self-antigens (29). As mentioned above, TLRs can also directly activate memory B cells for antibody production (17). Dectin-1 Dectin-1 is a member of the C-type lectin family of PRRs and recognizes b-glucans from fungal pathogens such as Candida albicans (30). Aside from the TLRs, Dectin-1 may be the PRR whose role in induction of adaptive immunity is best understood. Recent work has shown convincingly that Dectin-1 stimulation alone is sufficient to induce adaptive immunity (31). However, unlike TLRs, which induce Th1 responses, Dectin-1-induced adaptive immunity was shown to favor the differentiation of T cells into the Th17 phenotype (31, 32). Dectin-1 stimulation led to the preferential secretion of the Th17-supporting cytokine IL-23 in place of the Th1-skewing cytokine IL-12 (32). Notably, Th17 cells are specialized for defense against extracellular pathogens (33, 34). Thus, Dectin-1 triggering by extracellular fungi leads to a Th17 response resulting in recruitment of neutrophils, which engulf and kill the extracellular fungi leading to clearance of the infection. Dectin-1 is thus sufficient to induce an adaptive immune response that is tailored to the offending infection and is a perfect example of PRR-mediated tailoring of adaptive responses for the class of infection that is sensed. Furthermore, Dectin-1 signaling is critical for the clearance of fungal infections (35, 36). Interestingly, the signaling pathways used by Dectin-1 are very similar to those engaged by the antigen receptors in lymphocytes and are distinct from those used by TLRs (35, 37). This observation suggests that there are multiple signaling pathways that can lead to control of adaptive immunity. Dectin-1 recognizes b-glucans that are unique, to microbial non-self (30). Thus, barring discovery of a self-ligand for Dectin-1, Dectin-1 can reliably distinguish between self and non-self and control induction of adaptive immunity. Dectin-1, 2009 The Authors • Journal compilation 2009 Blackwell Munksgaard • Immunological Reviews 227/2009 225 Palm & Medzhitov Æ Innate instruction of adaptive immunity like TLRs, may also control the selective processing and presentation of contents from phagosomes containing fungal particles, although this has yet to be examined. Dectin-1, unlike the TLRs, is also a phagocytic receptor and can influence phagosome trafficking (38). This property may enhance the ability of Dectin-1 to induce selective presentation of non-self antigens. Finally, Dectin-1 engagement appears to be largely fungal specific. Thus, Dectin-1 can determine the class of infection and control the type of adaptive response induced accordingly (39). Cytosolic PRRs The cytosolic PRRs can be separated into two classes based on their mechanism of activation. The first class of cytosolic PRRs, which includes the founding nucleotide-binding oligomerization domain containing (Nod)-like receptor (NLR) family members Nod1 and Nod2, the retinoic acid-inducible gene I (RIG-I)-like helicases (RLHs), and presumably the IFN stimulatory DNA (ISD) sensor, directly detect cytosolic PAMPs and activate various signaling cascades (40, 41). The second class consists of members of the NLR family that are involved in the formation and activation of large, multimeric protein complexes referred to as inflammasomes, which control the activation of caspase-1, and the secretion of caspase-1-dependent cytokines such as IL-1b (42). At least some of these NLRs, such as Nacht domain-, LRR-, and PYD-containing protein 3 (NALP3), which can be activated by potassium efflux, sense activities that result from microbial infection rather than cytosolic PAMPs. However, other inflammasome NLRs, such as ICE protease-activating factor (Ipaf), are activated in response to cytosolic PAMPs. The role of cytosolic PRRs in control of adaptive immunity is just beginning to emerge. Cytosolic PRRs, unlike transmembrane PRRs, can distinguish between intracellular infections and extracellular infections, between cell-intrinsic infections and cell-extrinsic infections, and may even distinguish pathogenic microorganisms from harmless, commensal microorganisms. However, it is not clear whether or how cytosolic PRRs relay information about antigen origin to the adaptive immune system. We next discuss recent data on the role of cytosolic PRRs in control of adaptive immunity. NLRs Nod1 and Nod2 Nod1 and Nod2 sense bacterial infection through the detection of cytosolic peptidoglycan fragments from bacterial cell 226 walls. They activate an innate immune response that is critical for protection from bacteria that are able to escape the endolysosomal compartment and for production of antimicrobial peptides in the intestinal crypts (43–46). Notably, mutations in Nod2 have been linked to Crohn’s disease, causing great interest in the role of this PRR in immunity and pathology (45). In one study, Nod2-deficient animals had defects in antibody responses to protein immunization when the Nod2 ligand muramyldipeptide was used as adjuvant (47). Recent experiments have shown that specific activation of Nod1 in the absence of ligands for other PRRs is sufficient to induce a type 2 T- and B-cell response (48). Interestingly, this response was dependent upon Nod signaling in both hematopoietic and non-hematopoietic compartments. Furthermore, Nod1 stimulation was able to potentiate Th1, Th2, and Th17 responses initiated in conjunction with TLR ligands either during immunization or infection with Helicobacter pylori. It is thus suggested that, when given with TLR ligands, Nod1 potentiates and may shape adaptive immunity; however, because Nod1 stimulation potentiates all Th responses, it is unclear whether Nods are involved in controlling the type of response induced. However, because Nods can detect the presence of cell intrinsic, intracellular infections, it is tempting to speculate that they might be critically involved in shifting immunity in the intestine from tolerogenic to immunogenic (49). Furthermore, work in the human system has suggested that Nod stimulation specifically favors anti-bacterial Th17 responses through the selective induction of IL-23 and IL-1; this response was abrogated in cells from Crohn’s disease patients that carried a mutation in Nod2 (50). A role for the Nods in distinguishing pathogens from commensal organisms would fit well with the linkage between Nod2 mutations and Crohn’s disease as well as the ability of Nods to potentiate anti-microbial Th cell responses. However, further work will be required to solidify this hypothesis. NALP3 NALP3 inflammasomes can be activated in multiple ways. Experimentally, NALP3 inflammasomes are most often activated by pretreatment with lipopolysaccharide (LPS) followed by induction of potassium efflux via high dose adenosine triphosphate (51). Inflammasomes can also be activated by some intracellular Gram-positive pathogens by pore-forming exotoxins, as well as by the type III secretion system used by Gram-negative pathogens to inject virulence effectors into the host cell (49, 52). In each of these cases, in vitro activation of 2009 The Authors • Journal compilation 2009 Blackwell Munksgaard • Immunological Reviews 227/2009 Palm & Medzhitov Æ Innate instruction of adaptive immunity the NALP3 inflammasome in macrophages requires two signals- signal one (e.g. LPS) primes for activation and induces IL-1b production while signal two (e.g. potassium efflux) activates the inflammasome and leads to Caspase-1 activation and IL-1b processing and secretion. Although NALP3 is so far the best-studied NALP involved in inflammasome activation and IL-1b secretion, little is known about the involvement of NALP3 in control of adaptive immunity. The most compelling evidence for the involvement of NALP3 in activation of adaptive immunity comes from studies showing that NALP3 is required for priming of T-cell-dependent contact hypersensitivity responses to skin painting of haptens (53, 54). Recent studies have revealed that the common adjuvant Alum can also activate the NALP3 inflammasome, and one study suggests that NALP3 is required for optimal antibody responses following immunization using Alum as an adjuvant (55–58). It remains unclear whether inflammasome activation in vivo also always requires priming by TLRs, as it does in vitro. While the requirement for two separate signals to induce inflammasome activation may complicate experimental analysis, it may also provide the immune system with a powerful tool for sensing the insult responsible for disruption of homeostasis. As noted above, inflammasomes are specifically activated in response to bacteria that attempt to manipulate the host response through virulence factors, potentially providing a mechanism to distinguish between pathogens and commensal organisms. Thus, PAMP sensing alone by a transmembrane PRR might indicate the presence of microorganisms (commensal or pathogenic), while PAMP plus inflammasome activation might specifically indicate the presence of pathogenic microbes. One problem with this hypothesis is that inflammasomes can be activated under non-infectious conditions (e.g. in gout) in addition to their activation by pathogens (59). However, the inflammasomes that are activated by endogenous and/or non-infectious stimuli (e.g. uric acid crystals) may be largely specialized to deal with non-infectious stressors, and thus may not be connected to adaptive immunity. For example, ultraviolet treatment of keratinocytes also induces inflammasome activation (60). However, this activation may serve a very different purpose—it may induce mainly tissue repair rather than immunity. The triggering of the inflammasome by endogenous and/or non-infectious conditions could also be unintentional. Indeed, the endogenous triggers of NLRs are notable not for their normal role in maintaining homeostasis but instead for their role in pathological conditions such as gout (59). Cytosolic DNA sensor An innate immune response to cytosolic DNA leading to the production of large amounts of type I IFNs has recently been described (61, 62). Subsequently, the role of the cystolic DNA sensor, or interferon stimulatory DNA (ISD) sensor, in control of adaptive immunity has also been examined. DNA vaccines can induce both T-cell and antibody responses and activate both TLR9 and the yet-to-be-identified ISD sensor (63, 64). However, these two pathways can be distinguished via their signaling adapter usage. TLR9 signals through the TLR adapter myeloid differentiation factor 88 (MyD88), while the ISD sensor signals through TRAF family member-associated NF-jB activator (TANK)-binding kinase 1 (TBK1) to IFN regulatory factor 3 (IRF3) (65). Surprisingly, adaptive immune responses to DNA vaccines are largely TLR9 independent (64, 66). Instead, adaptive responses to DNA vaccines are TBK1- and type I IFN-dependent, implicating the ISD sensor (64). In these experiments, TBK1 was required in hematopoietic cells for CD4+ T-cell and B-cell responses, while TBK1 was required in non-hematopoietic cells for CD8+ T-cell responses. While the requirement for TBK1 for induction of adaptive immunity to DNA vaccines is compelling, the role of the ISD sensor in controlling adaptive immunity to infectious stimuli under physiological conditions and its ability to induce memory remain to be studied. Nonetheless, these experiments with DNA vaccines suggest that, in principle, the ISD sensor can control induction of antiviral T- and B-cell responses. It is particularly interesting to consider the mechanism by which the ISD sensor instructs the adaptive immune system on the origin of an antigen. Considering its cytosolic location, it is difficult to imagine a mechanism by which the coincidence of antigen and PAMP could be detected by the ISD sensor in such a way as to selectively induce responses to foreign antigens. However, it is certain that future studies will reveal more about the mechanisms of ISD-induced adaptive immunity. RIG-I ⁄ melanoma differentiation-associated gene 5 RIG-I and melanoma differentiation-associated gene 5 (MDA5) (which are both members of the RLH family) recognize RNA viruses in all cells, except for the specialized plasmacytoid DCs, and activate an antiviral innate immune response that is typified by the production of type I IFNs (67). The TLRs also play an important role in viral detection and defense, specifically in the IFN-producing plasmacytoid DC. Recent experiments examining the adaptive immune response 2009 The Authors • Journal compilation 2009 Blackwell Munksgaard • Immunological Reviews 227/2009 227 Palm & Medzhitov Æ Innate instruction of adaptive immunity to lymphocytic choriomeningitis virus (LCMV) and influenza infection in PRR-signaling adapter-deficient mice have begun to examine the relative importance of TLR versus RLH signaling in control of adaptive immunity. LCMV triggers type I IFN production through both TLR- and RLH-dependent pathways. CD8+ T cells mediate protective–adaptive immune responses to LCMV. Recent experiments demonstrated that the CD8+ T-cell response to LCMV was entirely TLR signaling dependent and that RLH signaling was dispensable for this response (68). Thus, in response to LCMV, TLRs are responsible for induction of protective CD8+ T-cell responses, and RIG-I ⁄ MDA5 signaling is insufficient to induce adaptive immunity. Both MyD88-dependent (TLR7) and IFN-b promoter stimulator 1 (IPS-1)-dependent (RIG-I ⁄ MDA5-dependent) pathways are induced in response to influenza A virus infection and mediate the innate response to the virus. Recently, the adaptive immune responses to influenza in MyD88-deficient and IPS-1-deficient mice were compared, revealing a role for RIG-I ⁄ MDA5 in control of adaptive immunity (69). In response to influenza A virus, IPS-1-dependent signaling (in the absence of TLR-dependent signaling) was sufficient to induce a CD8+ T-cell response. However, TLR-signaling was required for induction of CD4+ T-cell and antibody responses to influenza A; TLR signaling was required to provide protection from re-infection. Therefore, while RIG-I ⁄ MDA5 can induce CD8+ T-cell effector responses to certain viral infections, under these conditions they are insufficient to induce helper T-cell activation, antibody production, and protection from re-infection. It thus appears that adaptive immune responses to influenza A and LCMV are controlled largely by the TLRs. However, further research using additional viral infections must be performed before a final conclusion on the role of RIG-I ⁄ MDA5 in control of adaptive immunity can be reached. Coincident triggering of transmembrane and cytosolic PRRs Transmembrane and cytosolic PRRs differ notably in the types of microbes and infections that they detect. Transmembrane PRRs can sense the presence of both cell-intrinsic and non-cell intrinsic infections and are triggered by both pathogens and commensal organisms (70). Certain cytokines produced in response to infection, such as type I IFNs and TNF-a, also signal the presence of infection, and do not distinguish between cell-intrinsic and non-cell intrinsic infections, or pathogens and commensal organisms. In contrast, cytosolic PRRs are activated mainly by cell-intrinsic infections, and 228 some cytosolic PRRs may be specifically activated in response to pathogens and not commensal organisms (49). Thus, triggering of cytosolic PRRs may tag a cell as infected and may indicate pathogenicity. Because transmembrane PRRs, inflammatory cytokine receptors, and cytosolic PRRs are differentially activated in response to different types and classes of infections, these different signals probably serve distinct purposes. Isolated triggering versus coincident triggering of these different classes of receptors may lead to differing immune outcomes. Further studies into the role of different PRRs in immunity and the coincident triggering of intracellular PRRs, cytokine receptors, and cytosolic PRRs will certainly lead to a deeper understanding of the control of innate and adaptive immunity. PRRs in non-DCs and control of adaptive immunity The current view of innate instruction of adaptive immunity involves a linear progression of signals specifying to adaptive immune cells when and how to respond to a pathogenic insult. In this model, DCs are activated by the presence of infection through PRRs. Activated DCs presenting pathogenderived antigens migrate to the draining lymph node where they activate and instruct naive T cells. Finally, activated helper T cells migrate to the B-cell zone and provide antigen-specific help to B cells (8). However, a role for PRR signaling in non-DCs in control of adaptive immunity has been documented in a number of systems. For example, a role for PRRs on non-hematopoietic cells in control of adaptive immunity has been described for several systems (48, 64, 71, 72). Here, we focus on the role of TLRs on B cells in T-cell-dependent antibody responses. B-cell-intrinsic TLR signaling and control of T-dependent antibody responses Mice deficient in TLR signaling show severe defects in antibody responses to protein plus LPS in simple depot adjuvants. It was thought that T-cell help alone was sufficient to induce optimal T-dependent B-cell responses. Therefore, the defect in B-cell responses observed in mice deficient in TLR signaling could be expected to be entirely caused by the failure to induce a T-cell response. However, we recently demonstrated that TLR-dependent signals are required not only in DCs (to induce a T-cell response) but also in B cells themselves for induction of a robust T-dependent antibody response (21). This requirement for a B-cell-intrinsic TLR signal varied depending on the antibody isotype. IgM and IgG1 responses were largely TLR-dependent, IgG2c responses 2009 The Authors • Journal compilation 2009 Blackwell Munksgaard • Immunological Reviews 227/2009 Palm & Medzhitov Æ Innate instruction of adaptive immunity were nearly entirely TLR-dependent, and IgE responses were TLR-independent. Thus, it appears that TLRs on B cells themselves act as an additional checkpoint in the regulation of adaptive immune responses. Similar results were obtained in human B cells (27). It is interesting to speculate that one purpose of this additional checkpoint may be to regulate the magnitude and duration of the B-cell response based on the availability of TLR ligands in the lymph node. Presumably, the relative concentration of PRR ligands in the lymph node correlates with the extent of infection, and the absence of PRR ligands in the lymph node correlates with clearance of infection. In addition, this checkpoint may control effector versus memory cell differentiation; indeed, TLRs stimulation on B cells, which would represent persistence of infection and necessity for effector (plasma cell) generation, increases the expression of the critical regulator of plasma cell differentiation B-lymphocyte induced maturation protein-1 (Blimp-1) (21). Interestingly, TLR signaling in B cells is also critically involved in antibody-dependent autoimmunity (73–75). A number of investigators have since found similar requirements for TLRs on B cells in the induction of optimal antibody responses to various infections and immunizations. Notably, the link between TLR signaling on B cells and IgG2c production is maintained in every system examined, suggesting that this isotype is intimately connected to B-cell-intrinsic TLR signaling (73, 76–79). The reasons for this intimate connection and the need for TLR ligands in addition to T-cell help and the IgG2c switch factor IFN-c are unclear. However, it is tempting to hypothesize that the presence of B-cell stimulating TLR ligands in the lymph node might signal either the type or severity of the infection, and that this would shape the immune response to deal with the specific type of infection through production of IgG2c. Notably, IgG2 is highly efficient for both viral and bacterial clearance and efficiently fixes complement (80); therefore, TLRs on B cells may also shape the adaptive response to maximize resistance to the particular offending infection. Although many studies have found a role for TLRs in antibody responses, two reports have failed to find any role for TLRs in general, or on B cells in particular, in induction of antibody responses to common immunizations (79, 81, 82). Because the reason for this discrepancy in data sets was unclear, the role of B-cell-intrinsic TLR signals became controversial. However, we have recently found that this discrepancy is due to an experimental difference—experiments that found a role for TLRs were done using either native proteins in adjuvant or natural infections, while experiments finding little or no role for TLRs were done using proteins chemically modified by haptenation (haptenated proteins). We found that haptenated proteins, unlike native proteins, were uniquely immunogenic and thus could induce robust T- and B-cell responses in a TLR-independent manner (NP and RM, unpublished results). The reason for the unique immunogenicity of haptenated proteins is unclear, but it may be related to the antigen specificity of the adaptive responses that are induced - haptenated induce strong anti-hapten responses yet induce very weak responses to the carrier protein to which the haptens are conjugated. However, regardless of the reason for the immunogenicity of haptenated proteins, it is clear that haptenated proteins display altered innate requirements for induction of adaptive immunity that can affect the outcome of experiments testing adjuvant effects and often obscure the pathways used by common adjuvants. PRRs and adjuvanticity Common adjuvants contain at least two different types of activities that are critical for inducing adaptive immune responses to immunizations with soluble antigens. One activity is a depot activity that prevents the dispersion of soluble antigen and promotes the concentrated delivery of antigen to the draining lymph node where priming of the adaptive immune response can occur (83, 84). (Notably, infectious organisms do not require this activity, as they are naturally concentrated at the site of infection.) The other activity is based on activation of the innate immune system through triggering of appropriate PRRs, such as the TLRs, leading to activation of antigen-presenting cells and immunogenic presentation of antigen (85). The best adjuvants provide both activities and can convey immunogenicity on an otherwise non-immunogenic antigen. In the case of complete Freund’s adjuvant (CFA), emulsification in mineral oil acts largely to provide a depot, while heat killed mycobacteria act to trigger the innate immune system (83). Both depot and innate immune-stimulating activities are necessary for induction of robust adaptive immune responses to soluble proteins (83). Some common adjuvants, such as CFA, contain components that have both activities, while other common adjuvants have only one of the two necessary activities. These adjuvants are, by themselves, insufficient to induce robust adaptive responses. For example, TLR ligands, which efficiently stimulate the innate immune system, are insufficient to induce adaptive immunity when used alone as adjuvant for soluble proteins (86). 2009 The Authors • Journal compilation 2009 Blackwell Munksgaard • Immunological Reviews 227/2009 229 Palm & Medzhitov Æ Innate instruction of adaptive immunity Fig. 2. Proteins commonly used for immunizations are highly contaminated with TLR ligands. Bone marrow-derived DCs (BMDCs) from wildtype or myeloid differentiation factor 88 (MyD88) ⁄ TIRdomain-containing adapter-inducing interferon-b (TRIF)-deficient mice were stimulated with lipopolysaccharide (LPS) (100 ng ⁄ ml), ovalbumin (OVA), endotoxin-free human serum albumin (HSA), keyhole limpet hemocyanin (KLH), or chicken c-globulin (CGG) for 20 h before measuring interleukin-6 (IL-6) secretion into the supernatant by enzymelinked immunosorbent assay. All proteins were used at 300 lg ⁄ ml. This failure to induce adaptive immunity is largely caused by the lack of depot adjuvant, and thus the rapid dispersion of the soluble antigen such that the required local concentration of antigen and PAMP is never reached (83, 86). By contrast, Alum and incomplete Freund’s adjuvant (IFA) are efficient depot adjuvants but contain minimal amounts of PRR (84). For this reason, immunizations with purified proteins, such as endotoxin-free human serum albumin (which is free of contaminating TLR ligands or other PRR ligands) (Fig. 2) or subunit vaccine formulations, in either Alum or IFA fail to induce robust adaptive responses because of the lack of a sufficient PRR stimulating activity (13, 21, 87, 88). However, the addition of a single TLR ligand, such as LPS, to a mixture of depot adjuvant and soluble protein allows robust induction of adaptive immunity (13, 21). Most proteins that are commonly used for immunization studies- including keyhole limpet hemocyanin, bovine serum albumin, ovalbumin, and chicken c-globulin- are highly contaminated with PRR ligands (Fig. 2). These contaminating PAMPs provide the necessary PRR stimulating activity, which accounts for the induction of adaptive immune response by contaminated proteins in depot adjuvants, such as IFA and Alum. The most common contaminants are LPS and bacterial lipoproteins; thus, the immunogenic effects of these contaminants are dependent upon TLR signaling. However, PAMP contaminants are not necessarily restricted to TLR ligands. For example, recombinant proteins may also be contaminated with TLR-independent PAMPs, such as Nod ligands. Also, we 230 have recently observed that, in contrast to native, PAMP-free proteins, PAMP-free haptenated proteins in IFA or Alum induce robust adaptive immune responses (NP and RM, unpublished results). Thus, certain chemical modifications of antigens can also influence the perceived requirements for innate immunostimulatory adjuvants. Failure to consider these possibilities when performing immunization-based experiments can greatly confuse the interpretation of data and lead to incorrect conclusions on the requirements for innate instruction of adaptive immunity. Ideally, experiments testing the role of a particular PAMP or PRR should be performed in a system where the role of a given PRR can be isolated from the effects of other PRRs. This separation can be achieved via targeting of a particular PRR using its corresponding PAMP in conjunction with PRR-free protein antigens in PRR-free depot adjuvants. In particular, it is critical that experiments testing the sufficiency of a PRR for induction of adaptive immunity be performed in a manner where the role of a single PRR can be sufficiently isolated. In the absence of this type of experiment, it is impossible to know whether a PRR is sufficient for activation of adaptive immunity. An additional activity that adjuvants may provide relates to the requirement for antigen and PRR to be delivered concomitantly and, ideally, to be delivered to the same phagosome, to induce a maximal response. Adjuvants, such as Alum and IFA, may thus also act to deliver antigen and PAMP in a complex that results in delivery to the same phagosome. Indeed, the co-delivery of antigen and PAMP is critical for induction of adaptive immunity; separate immunizations with PAMP and antigen emulsified separately fail to induce robust adaptive immune responses despite the presence of both necessary adjuvant activities (11). Furthermore, the immunogenicity of a protein can be increased via conjugation with a PAMP, as is achieved in the case of flagellin-fusion proteins (89). It is interesting to consider how the ability of a protein to complex with PAMPs may contribute to the immunogenicity and ⁄ or immunodominance of certain proteins over others. Common adjuvants also probably have other, yet to be discovered activities, which may or may not be dependent upon classical PRRs. For example, IFA can induce lymph node swelling in a TLR-independent manner (NP and RM, unpublished observations); the mechanism responsible for this innate response to IFA has yet to be defined. It is also of interest to note that Alum has recently been shown to cause IL-1b secretion through activation of the NALP3 inflammasome, which may account for Alum’s adjuvanticity in addition to its depot effect (55–58). 2009 The Authors • Journal compilation 2009 Blackwell Munksgaard • Immunological Reviews 227/2009 Palm & Medzhitov Æ Innate instruction of adaptive immunity When is adaptive immunity induced? While much has been learned over the past decade about innate instruction of adaptive immunity, a number of fundamental questions still remain. For example, it is not entirely clear what determines whether an adaptive immune response will be induced during a given infection. A textbook notion is that during an infection an innate immune response is first induced and attempts to eliminate the pathogen, and if it is not sufficient to clear the infection, then the adaptive immune response follows. This notion however is incompatible with the current understanding of the mechanisms of initiation of adaptive immune responses. Namely, tissueresident DCs function as sensors of microbial invaders, and upon contact with pathogens, they mature and migrate to the lymph nodes where they activate naive T cells. According to this view, regardless of the ability of the innate immune system to contain infection, DCs that have encountered microbes should induce an adaptive immune response. However, lowgrade asymptomatic infections are commonplace, and they are unlikely to always result in the activation of adaptive immune responses. It is not clear whether activation of the adaptive immune response is simply a matter of pathogen load, which would imply the existence of a threshold for activation of adaptive immunity, or whether additional mechanisms exist that determine when an adaptive immune response is induced based on other, currently unknown, parameters of infection. Conclusions Many recent studies have highlighted and clarified the role of PRRs in induction and instruction of adaptive immunity and have revealed roles for new PRRs, and new roles for old PRRs, in control of adaptive immunity. We now know that PRRinduced signals control the adaptive immune system at various checkpoints controlling the activation, effector class, magnitude, duration, and memory of an adaptive immune response, and that the ability of a PRR to control adaptive immunity at these checkpoints depends upon the ability of that PRR to define the ongoing infection and relay that information to the adaptive immune system. While the roles of various PRRs in control of adaptive immunity are becoming clearer, it is also certain that we are far from a complete understanding of how innate immunity shapes adaptive responses to maximize defense and the specific role that each class of PRRs plays in this process. Future studies will undoubtedly reveal new mechanisms of innate control of adaptive immunity. References 1. Hoffmann JA, Kafatos FC, Janeway CA, Ezekowitz RA. Phylogenetic perspectives in innate immunity. Science 1999;284:1313–1318. 2. Medzhitov R, Janeway CA Jr. Innate immunity: the virtues of a nonclonal system of recognition. Cell 1997;91: 295–298. 3. Cooper MD, Alder MN. The evolution of adaptive immune systems. Cell 2006; 124:815–822. 4. Janeway CA Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 1989; 54:1–13. 5. Gallegos AM, Bevan MJ. Central tolerance: good but imperfect. Immunol Rev 2006; 209:290–296. 6. Macian F, Im SH, Garcia-Cozar FJ, Rao A. T-cell anergy. Curr Opin Immunol 2004;16:209–216. 7. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell 2008;133:775–787. 8. Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol 2004;5: 987–995. 9. Carroll MC. The complement system in regulation of adaptive immunity. Nat Immunol 2004;5:981–986. 10. Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science 2004;304: 1014–1018. 11. Blander JM, Medzhitov R. On regulation of phagosome maturation and antigen presentation. Nat Immunol 2006;7: 1029–1035. 12. Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature 2006;440: 808–812. 13. Pasare C, Medzhitov R. Toll-dependent control mechanisms of CD4 T-cell activation. Immunity 2004;21:733–741. 14. Sporri R, Reis e Sousa C. Inflammatory mediators are insufficient for full dendritic cell activation and promote expansion of CD4+ T-cell populations lacking helper function. Nat Immunol 2005;6:163–170. 15. Reiner SL. Development in motion: helper T cells at work. Cell 2007;129:33–36. 16. Badovinac VP, Porter BB, Harty JT. Programmed contraction of CD8(+) T cells after infection. Nat Immunol 2002;3:619–626. 17. Bernasconi NL, Traggiai E, Lanzavecchia A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science 2002;298:2199–2202. 18. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006;124:783–801. 19. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature 1998;392:245–252. 20. Pasare C, Medzhitov R. Toll-like receptors: linking innate and adaptive immunity. Adv Exp Med Biol 2005;560:11–18. 21. Pasare C, Medzhitov R. Control of B-cell responses by Toll-like receptors. Nature 2005;438:364–368. 22. Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol 2001; 2:947–950. 23. Yarovinsky F, Kanzler H, Hieny S, Coffman RL, Sher A. Toll-like receptor recognition regulates immunodominance in an antimicrobial CD4+ T-cell response. Immunity 2006;25:655–664. 24. West MA, et al. Enhanced dendritic cell antigen capture via toll-like receptor-induced 2009 The Authors • Journal compilation 2009 Blackwell Munksgaard • Immunological Reviews 227/2009 231 Palm & Medzhitov Æ Innate instruction of adaptive immunity 25. 26. 27. 28. 29. 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. actin remodeling. Science 2004;305: 1153–1157. Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+ CD25+ T cell-mediated suppression by dendritic cells. Science 2003;299:1033–1036. Vella AT, Dow S, Potter TA, Kappler J, Marrack P. Cytokine-induced survival of activated T cells in vitro and in vivo. Proc Natl Acad Sci USA 1998;95:3810–3815. Ruprecht CR, Lanzavecchia A. Toll-like receptor stimulation as a third signal required for activation of human naive B cells. Eur J Immunol 2006;36: 810–816. Nagai Y, et al. The radioprotective 105 ⁄ MD-1 complex links TLR2 and TLR4 ⁄ MD-2 in antibody response to microbial membranes. J Immunol 2005;174:7043–7049. Herlands RA, Christensen SR, Sweet RA, Hershberg U, Shlomchik MJ. T cell-independent and Toll-like receptor-dependent antigen-driven activation of autoreactive B cells. Immunity 2008;29:249–260. Brown GD, Gordon S. Immune recognition. A new receptor for beta-glucans. Nature 2001;413:36–37. LeibundGut-Landmann S, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol 2007;8:630–638. Acosta-Rodriguez EV, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol 2007;8:639–646. Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol 2007;25: 821–852. Mangan PR, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 2006;441:231–234. Gross O, et al. Card9 controls a non-TLR signalling pathway for innate anti-fungalimmunity. Nature 2006;442:651–656. Taylor PR, et al. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat Immunol 2007;8: 31–38. Rogers NC, et al. Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity 2005;22:507–517. Herre J, et al. Dectin-1 uses novel mechanisms for yeast phagocytosis in macrophages. Blood 2004;104: 4038–4045. Dennehy KM, Brown GD. The role of the beta-glucan receptor Dectin-1 in control of fungal infection. J Leukoc Biol 2007;82:253–258. 232 40. Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature 2006;442:39–44. 41. Takeuchi O, Akira S. Recognition of viruses by innate immunity. Immunol Rev 2007; 220:214–224. 42. Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell 2004;117:561–574. 43. Fritz JH, Ferrero RL, Philpott DJ, Girardin SE. Nod-like proteins in immunity, inflammation and disease. Nat Immunol 2006;7: 1250–1257. 44. Girardin SE, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 2003;300:1584– 1587. 45. Cho JH. The genetics and immunopathogenesis of inflammatory bowel disease. Nat Rev Immunol 2008;8:458–466. 46. Girardin SE, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 2003;278:8869–8872. 47. Kobayashi KS, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 2005;307:731–734. 48. Fritz JH, et al. Nod1-mediated innate immune recognition of peptidoglycan contributes to the onset of adaptive immunity. Immunity 2007;26:445–459. 49. Kaparakis M, Philpott DJ, Ferrero RL. Mammalian NLR proteins; discriminating foe from friend. Immunol Cell Biol 2007; 85:495–502. 50. van Beelen AJ, et al. Stimulation of the intracellular bacterial sensor NOD2 programs dendritic cells to promote interleukin-17 production in human memory T cells. Immunity 2007;27:660–669. 51. Mariathasan S, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006;440:228–232. 52. Ye Z, Ting JP. NLR, the nucleotide-binding domain leucine-rich repeat containing gene family. Curr Opin Immunol 2008;20:3–9. 53. Sutterwala FS, et al. Critical role for NALP3 ⁄ CIAS1 ⁄ cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 2006;24:317– 327. 54. Watanabe H, et al. Activation of the IL1beta-processing inflammasome is involved in contact hypersensitivity. J Invest Dermatol 2007;127:1956–1963. 55. Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 2008;453:1122–1126. 56. Kool M, et al. Cutting Edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J Immunol 2008;181:3755–3759. 57. Franchi L, Nunez G. The Nlrp3 inflammasome is critical for aluminium hydroxidemediated IL-1beta secretion but dispensable for adjuvant activity. Eur J Immunol 2008; 38:2085–2089. 58. Li H, Willingham SB, Ting JP, Re F. Cutting edge: inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. J Immunol 2008;181:17–21. 59. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006;440:237–241. 60. Keller M, Ruegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell 2008;132: 818–831. 61. Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 2006;24:93–103. 62. Ishii KJ, et al. A Toll-like receptor-independent antiviral response induced by doublestranded B-form DNA. Nat Immunol 2006; 7:40–48. 63. Gurunathan S, Klinman DM, Seder RA. DNA vaccines: immunology, application, and optimization*. Annu Rev Immunol 2000;18: 927–974. 64. Ishii KJ, et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 2008;451: 725–729. 65. Ishii KJ, Akira S. Innate immune recognition of, and regulation by, DNA. Trends Immunol 2006;27:525–532. 66. Spies B, et al. Vaccination with plasmid DNA activates dendritic cells via Toll-like receptor 9 (TLR9) but functions in TLR9-deficient mice. J Immunol 2003;171:5908–5912. 67. Takeuchi O, Akira S. MDA5 ⁄ RIG-I and virus recognition. Curr Opin Immunol 2008;20: 17–22. 68. Jung A, et al. Lymphocytoid choriomeningitis virus activates plasmacytoid dendritic cells and induces a cytotoxic T-cell response via MyD88. J Virol 2008;82:196–206. 69. Koyama S, et al. Differential role of TLR- and RLR-signaling in the immune responses to influenza A virus infection and vaccination. J Immunol 2007;179:4711–4720. 70. Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature 2007;449:819–826. 71. Sato A, Iwasaki A. Induction of antiviral immunity requires Toll-like receptor signaling in both stromal and dendritic cell compartments. Proc Natl Acad Sci USA 2004; 101:16274–16279. 2009 The Authors • Journal compilation 2009 Blackwell Munksgaard • Immunological Reviews 227/2009 Palm & Medzhitov Æ Innate instruction of adaptive immunity 72. Fritz JH, Le Bourhis L, Magalhaes JG, Philpott DJ. Innate immune recognition at the epithelial barrier drives adaptive immunity: APCs take the back seat. Trends Immunol 2008;29:41–49. 73. Groom JR, et al. BAFF and MyD88 signals promote a lupuslike disease independent of T cells. J Exp Med 2007;204: 1959–1971. 74. Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Tolllike receptors. Nature 2002;416:603–607. 75. Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol 2006;6:823–835. 76. Guay HM, Andreyeva TA, Garcea RL, Welsh RM, Szomolanyi-Tsuda E. MyD88 is required for the formation of long-term humoral immunity to virus infection. J Immunol 2007;178:5124–5131. 77. Heer AK, et al. TLR signaling fine-tunes antiinfluenza B cell responses without regulating 78. 79. 80. 81. 82. 83. effector T-cell responses. J Immunol 2007; 178:2182–2191. Jegerlehner A, Maurer P, Bessa J, Hinton HJ, Kopf M, Bachmann MF. TLR9 signaling in B cells determines class switch recombination to IgG2a. J Immunol 2007;178: 2415–2420. Meyer-Bahlburg A, Khim S, Rawlings DJ. B cell intrinsic TLR signals amplify but are not required for humoral immunity. J Exp Med 2007;204:3095–3101. Hinton HJ, Jegerlehner A, Bachmann MF. Pattern recognition by B cells: the role of antigen repetitiveness versus Toll-like receptors. Curr Top Microbiol Immunol 2008;319:1–15. Gavin AL, et al. Adjuvant-enhanced antibody responses in the absence of toll-like receptor signaling. Science 2006;314:1936–1938. Nemazee D, Gavin A, Hoebe K, Beutler B. Immunology: Toll-like receptors and antibody responses. Nature 2006;441:E4. Billiau A, Matthys P. Modes of action of Freund’s adjuvants in experimental models of 84. 85. 86. 87. 88. 89. autoimmune diseases. J Leukoc Biol 2001;70:849–860. Lindblad EB. Aluminium compounds for use in vaccines. Immunol Cell Biol 2004;82: 497–505. Kaisho T, Akira S. Toll-like receptors as adjuvant receptors. Biochim Biophys Acta 2002; 1589:1–13. Klinman DM. Adjuvant activity of CpG oligodeoxynucleotides. Int Rev Immunol 2006;25:135–154. Guy B. The perfect mix: recent progress in adjuvant research. Nat Rev Microbiol 2007;5:505–517. Pulendran B, Ahmed R. Translating innate immunity into immunological memory: implications for vaccine development. Cell 2006;124:849–863. Cuadros C, Lopez-Hernandez FJ, Dominguez AL, McClelland M, Lustgarten J. Flagellin fusion proteins as adjuvants or vaccines induce specific immune responses. Infect Immun 2004;72: 2810–2816. 2009 The Authors • Journal compilation 2009 Blackwell Munksgaard • Immunological Reviews 227/2009 233