Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Structural alignment wikipedia , lookup
Degradomics wikipedia , lookup
Protein design wikipedia , lookup
List of types of proteins wikipedia , lookup
Circular dichroism wikipedia , lookup
Protein domain wikipedia , lookup
Protein folding wikipedia , lookup
Homology modeling wikipedia , lookup
Bimolecular fluorescence complementation wikipedia , lookup
Protein structure prediction wikipedia , lookup
Protein moonlighting wikipedia , lookup
Protein purification wikipedia , lookup
Intrinsically disordered proteins wikipedia , lookup
Western blot wikipedia , lookup
Nuclear magnetic resonance spectroscopy of proteins wikipedia , lookup
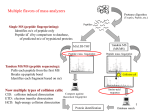
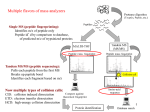
BIOINFORMATICS APPLICATIONS NOTE Vol. 25 no. 15 2009, pages 1980–1981 doi:10.1093/bioinformatics/btp301 Gene expression A toolbox for validation of mass spectrometry peptides identification and generation of database: IRMa Véronique Dupierris1 , Christophe Masselon2 , Magali Court2 , Sylvie Kieffer-Jaquinod2 and Christophe Bruley2,∗ Rhône-Alpes Futur, 89 rue Bellecombe, 69003 Lyon and 2 CEA, DSV, iRTSV, Laboratoire d’Etude de la Dynamique des Protéomes, INSERM, U880, Université Joseph Fourier, Grenoble F-38054, France 1 Fondation Received on December 5, 2008; revised and accepted on April 30, 2009 Advance Access publication May 6, 2009 Associate Editor: John Quackenbush ABSTRACT Summary: The IRMa toolbox provides an interactive application to assist in the validation of Mascot search results. It allows automatic filtering of Mascot identification results as well as manual confirmation or rejection of individual PSM (a match between a fragmentation mass spectrum and a peptide). Dynamic grouping and coherence of information are maintained by the software in real time. Validated results can be exported under various forms, including an identification database (MSIdb). This allows biologists to compile search results from a whole study in a unique repository in order to provide a summarized view of their project. IRMa also features a fully automated version that can be used in a high-throughput pipeline. Given filter parameters, it can delete hits with no significant PSM, regroup hits identified by the same peptide(s) and export the result to the specified format without user intervention. Availability: http://biodev.extra.cea.fr/docs/irma (java 1.5 or higher needed) Contact: [email protected] 1 INTRODUCTION The Mascot search engine uses mass spectrometry data to identify proteins by matching experimental and theoretical peptide mass spectra (Aebersold and Mann, 2003). However, it is necessary to eliminate false matches (Baldwin, 2004; Nesvizhskii and Aebersold, 2004) in order to avoid incorrect protein identification. The Mascot html output is not designed for this kind of validation, and exporting the data to Excel for subsequent validation results in loss of data consistency. Moreover, validation being the first step in data interpretation, subsequent data mining often requires compilation of numerous search results. IRMa is a validation toolbox for Mascot identification results that allow users to validate and export the identified proteins and their PSM to various formats. Its main originality is to filter matches rather than identified proteins. 2 VALIDATING HITS VERSUS PSM For each submitted spectrum (query), Mascot attempts to match up to 10 peptides from a sequence database. A score, based on the ∗ To whom correspondence should be addressed. 1980 absolute probability that the observed match is a random event, is assigned to each peptide spectrum matches (PSMs). For each query, PSMs are ranked according to their score. Mascot then groups PSMs into protein hits. A hit contains not only all proteins covered by the same set of PSMs but also all proteins covered by a subset of these PSMs. A hit score, based on its matching PSM scores, is assigned to each protein. While existing tools (Bouyssié et al., 2007) validate whole hits, selecting confident PSMs and rejecting dubious ones undoubtedly allow more accurate identification. When validating whole hits, false PSMs can still be considered in the result. On the other hand, validation at the PSM level affects the grouping consistency (and hits properties such as score and coverage): rejecting PSM without grouping update leads to redundancy of the protein hits’ list. IRMa allows validation at the PSM level, while circumventing the associated difficulties. 3 FEATURES IRMa uses the Mascot Parser distributed free of charge by Matrix Science to build an identification result from the native ‘.dat’ files generated by the Mascot server. The parser can be invoked with different report parameters, such as the number of hits or significance thresholds. At this stage, the same information and grouping shown in the Mascot result page are available but displayed in a structured and accessible layout by IRMa (see Fig. 1). PSMs relevant for protein identifications are flagged as significant. A PSM having the same sequence as a significant one but associated to another query and with a lower score are flagged as duplicated. For validation purposes, in addition to the significant and duplicated categories, IRMa introduces a third category in which ambiguous PSMs are classified. This third category regroups PSMs rejected by filtering and that do not contribute to protein identification. While PSMs’ classification into these categories can be done manually, several filters applied on the Mascot parser results are proposed. They provide rules to assign PSMs to significant or ambiguous categories and can be based on PSM ranks, scores, on mandatory post-translational modifications or on PSMs properties expression. This automatic classification can always be adjusted; the user can restore PSMs from the ambiguous group to the significant group or vice versa. Nevertheless, IRMa will ensure that the exact same sequence cannot appear twice © The Author 2009. Published by Oxford University Press. All rights reserved. For Permissions, please email: [email protected] [18:08 26/6/2009 Bioinformatics-btp301.tex] Page: 1980 1980–1981 IRMa 4 Fig. 1. IRMa snapshot. User can browse the submitted spectra (queries) (1) or the identified protein hits and PSMs (2). The main component of the interface displays query statistics or, as shown here, hit information. In this case, the upper part (3) shows the master protein description and properties together with the lists of same-set and sub-set proteins. PSMs classified in significant, ambiguous and duplicated categories are displayed for the current hit (4). Biological studies often require the comparison of numerous samples. Furthermore, each sample can be fractionated before analysis to generate several aliquots, which in turn result in as many Mascot searches. One of the major challenges facing the biologist resides in the high redundancy in identification lists. Compiling multiple search results through multiple Excel files or html results pages is impractical. In order to overcome this difficulty, the creation of a unique database, containing the identification results of a whole project, allows the user to access information on all analyzed samples at once. IRMa was used in our laboratory to compile data from a largescale proteomics study of A. thaliana chloroplasts. The MSIdb was compiled in a fully unattended mode after providing the software with filter parameters and an output path. The database was populated using search results from ∼585 000 Mascot queries generated from ∼500 LC–MS/MS analyses, with minimal user intervention. 5 in the significant category. Dynamic classification has an impact on protein hits: when a PSM is declared ambiguous, it becomes irrelevant to proteins identification. IRMa takes into account these changes to ensure consistency of information such as protein coverage and identification score and dynamically proposes new protein hit sets and sub-sets based on significant PSMs. For instance, two proteins of a same hit that initially shared only a sub-set of PSMs can become indistinguishable (all significant PSMs are shared) if specific PSMs are judged ambiguous. Another possibility is the merge of two distinct hits into a unique one if discriminating PSMs are flagged as ambiguous. For example, given the following two hits, ACTG_HUMAN {pep_63, pep_133, pep_167, pep_254, pep_311} and Q53G99_HUMAN {pep_63, pep_133,pep_139, pep_254, pep_311}, if PSM pep_139 is declared ambiguous, IRMa will propose grouping Q53G99_HUMAN and ACTG_HUMAN as the same hit. The user can validate new grouping proposed by the application by merging hits that become sub-sets or same sets of another and by deleting hits that no longer contain any significant PSM. This implies that hits are reordered according to their scores. The validated result can finally be exported under various forms: to the widely used Microsoft Excel format; to a PDF format including graphical representation of annotated spectra1 or to MSIdb2 . The information recorded in all exports can be customized. For instance, in Microsoft Excel format, user can choose which worksheets (protein summary, similar proteins, peptide list, etc.) and which columns will be present. Information on Mascot search parameters or automatic filtering is also reported in the output file. The MSIdb stores this information as well as that on grouping and matches between proteins and PSMs. Exporting to MSIdb is especially suitable for large-scale studies. 1 2 LARGE-SCALE STUDIES PERSPECTIVES IRMa allows generation of an MSIdb for a given project. To enhance data mining of comprehensive identification catalog, it would therefore be helpful to integrate identification databases with information from other data sources (e.g. web-based public repositories). We are currently working on a tool to achieve this aim and to further investigate identified proteins. Grouping identifications at the whole database level to remove redundancy among samples or comparing multiple experiments are some of the available operations. ACKNOWLEDGEMENTS We thank Matrix Science technical support team for their timely answers to our requests, the IRMa workgroup for their help in application specification and design, and all EDyP Laboratory team members for testing and feedback. Funding: CEA, INSERM and the Rhône-Alpes Génopôle. Conflict of Interest: none declared. REFERENCES Aebersold,R. and Mann,M. (2003) Mass spectrometry-based proteomics. Nature, 422, 198–207. Baldwin,M. (2004) Protein identification by mass spectrometry: issues to be considered. Mol. Cell. Proteomics 3, 1–9. Bouyssié,D. et al. (2007) MFPaQ, a new software to parse, validate, and quantify proteomic data generated by ICAT and SILAC mass spectrometric analyses: application to the proteomic study of membrane proteins from primary human endothelial cells. Mol. Cell. Proteomics 6, 1621–1637. Nesvizhskii,A.I. and Aebersold,R. (2004) Analysis, statistical validation and dissemination of large-scale proteomics datasets generated by tandem MS. Drug Discov. Today, 9, 173–181. MCP guideline compliant - for single peptide hits and PTM peptides hits. For database structure see supplementary material. 1981 [18:08 26/6/2009 Bioinformatics-btp301.tex] Page: 1981 1980–1981